A novel role for CDK2 as a new approach for treating neutrophilic inflammation
08-22-2019
Deng lab research published in the Proceedings of the National Academy of Sciences
Neutrophils are the first cells recruited to an immune stimulus stemming from infection. Its migration is essential for inflammatory responses to kill pathogens. However, overamplified or chronic neutrophil recruitment directly leads to autoimmune diseases such as rheumatic arthritis, diabetes, neurodegenerative diseases, and cancer.
To discover novel therapeutic targets which can fine tune neutrophil-mediated inflammation, the Deng lab initiated a screen on neutrophil-specific microRNAs – a regulator of transcription. They identified that miR-199 overexpression can hinder acute neutrophil recruitment to inflammatory sites and relieve systemic inflammation without compromising overall immune functions.
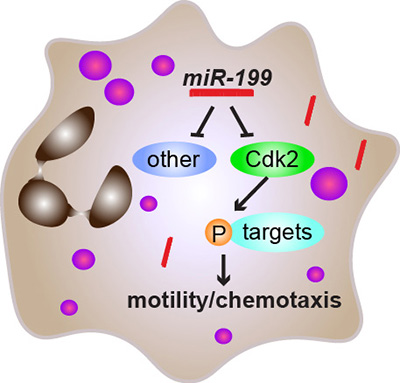
Subsequent identification of miR-199 regulated targets found that it supresses the cell cycle-related gene, CDK2. CDK2 is well known for its role in regulating a cell’s life cycle facilitating DNA replication, however, its link to neutrophil migration or function was previously unknown. Through their study, they discovered that inhibiting CDK2 but not DNA replication, disrupts neutrophil migration without inducing cell death.
These findings present new avenues to alleviate systemic inflammation and a previously unknown role for CDK2 outside classical functions. The study is ongoing, and the following step is to understand the detailed molecular mechanisms for how CDK2 suppresses neutrophil migration and lethal inflammation.
Full citation: Alan Y. Hsu, Decheng Wang, Sheng Liu, Justice Lu, Ramizah Syahirah, David A. Bennin, Anna Huttenlocher, David M. Umulis, Jun Wan, and Qing Deng (2019) Phenotypical microRNA screen reveals a noncanonical role of CDK2 in regulating neutrophil migration. DOI: https://doi.org/10.1073/pnas.1905221116. PNAS September 10, 2019 116 (37) 18561-18570
Contact: Qing Deng
ABSTRACT
Phenotypical microRNA screen reveals a noncanonical role of CDK2 in regulating neutrophil migration
Alan Y. Hsu, Decheng Wang, Sheng Liu, Justice Lu, Ramizah Syahirah, David A. Bennin, Anna Huttenlocher, David M. Umulis, Jun Wan, and Qing Deng
Neutrophil migration is essential for inflammatory responses to kill pathogens; however, excessive neutrophilic inflammation also leads to tissue injury and adverse effects. To discover novel therapeutic targets that modulate neutrophil migration, we performed a neutrophil-specific microRNA (miRNA) overexpression screen in zebrafish and identified 8 miRNAs as potent suppressors of neutrophil migration. Among those, miR-199 decreases neutrophil chemotaxis in zebrafish and human neutrophil-like cells. Intriguingly, in terminally differentiated neutrophils, miR-199 alters the cell cycle-related pathways and directly suppresses cyclin-dependent kinase 2 (Cdk2), whose known activity is restricted to cell cycle progression and cell differentiation. Inhibiting Cdk2, but not DNA replication, disrupts cell polarity and chemotaxis of zebrafish neutrophils without inducing cell death. Human neutrophil-like cells deficient in CDK2 fail to polarize and display altered signaling downstream of the formyl peptide receptor. Chemotaxis of primary human neutrophils is also reduced upon CDK2 inhibition. Furthermore, miR-199 overexpression or CDK2 inhibition significantly improves the outcome of lethal systemic inflammation challenges in zebrafish. Our results therefore reveal previously unknown functions of miR-199 and CDK2 in regulating neutrophil migration and provide directions in alleviating systemic inflammation.