Aguilar Lab - "Use of induced pluripotent stem cells from Lowe syndrome patients provide insight into disease mechanism"
04-02-2018
Lowe Syndrome (LS) is a devastating genetic disease characterized by abnormalities in the eyes, brain and kidneys that unfortunately leads to the premature death of affected children due to renal failure. Despite being described more than 60 years ago, this condition lacks a clear delineation of its mechanism and no specific cure is available. One contributing cause to this slow progress has been the absence of proper disease models for this condition and the inaccessibility of patient cells from the major affected organs.
However, using patient skin fibroblasts the Aguilar lab recently reported the first successful preparation of Lowe syndrome induced Pluripotent Stem Cells (iPSCs) and their reprogramming as renal cells1. This work not only represents a technological advance for the LS research field, but also provided insight as to how the patient’s kidney complications develop.
On the one hand, this work constituted the first application of iPSC/reprogramming technology to LS, opening the possibility of in vitro generation of cell types difficult to obtain from patients (e.g., brain and kidney) and of more sophisticated disease models such as in vitro-generated organoids. Importantly, this study also sets up the basis for future cell replacement therapies.
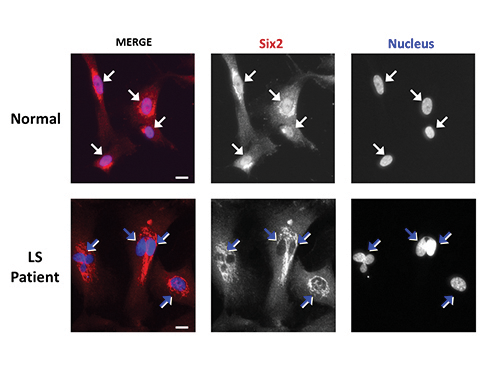
On the other hand, monitoring the process of in vitro kidney cell differentiation provided clues as to how renal deficiency arises in patients. Specifically, Aguilar lab graduate student Wen-Chieh Hsieh and colleagues found that in LS kidney cells the transcription factor Six2 (crucial for renal development) was abnormally retained outside the cell nucleus (Fig. 1) impairing its gene regulatory function. This deficient Six2 activity caused decreased production of the so-called proximal tubular cells which are involved in critical functions of the kidney, such as avoiding the excretion of important serum proteins. Therefore, two important implications arise from these findings:
-
Tubular cells are less readily available within LS kidney cell populations. This observation supports the hypothesis that LS patients experience kidney developmental abnormalities, particularly affecting tubular cells. Indeed, it is well-known that LS patients display deficient tubular function and tubular atrophy. Further, since Six2 is also involved in craniofacial and eye development as well as in neuro-protection, it is possible that affected function or regulation of this transcription factor contributes to characteristic phenotypes and symptoms of LS in other tissues or organs.
-
Patients would have difficulties to replenish tubular cells following wear or injury. In fact, there are reports of progressive tubular function loss in LS patients. A body of evidence collected by many groups indicate the existence of kidney-localized progenitor cells able to differentiate into tubular cells when needed to maintain renal functionality. The results presented in this study suggest that the availability of such cell replenishing pool is compromised in LS patients.
Misregulation of a differentiation pathway is a novel LS phenotype that is predicted to have great impact in patients’ renal function. Further, this work suggests that developing strategies directed to enhance proper Six2 function or to prevent its retention outside the nucleus constitute viable options to maintain renal function in LS patients.
Reference
1. Hsieh W-C, Ramadesikan S, Fekete D. and Aguilar RC. Kidney-differentiated cells derived from Lowe Syndrome patient’s iPSCs show Ciliogenesis defects and Six2 retention at the Golgi complex. PLOS ONE. 2018 Feb 14;13(2):e0192635. doi: 10.1371/journal.pone.0192635.
This work was supported by the National Institutes of Health and the Lowe Syndrome Trust under Grants 1R01DK109398-01 and BU/CO/2014 to R. C. Aguilar.