In Silico drug discovery method for computationally modelled target proteins
08-18-2016
Traditionally, drug molecules have been identified through high-throughput experimental screening. Since this process is costly, a computational method called Virtual Screening (VS) has emerged as a complementary approach since the early 1990s. VS can be performed effectively if the tertiary structure of the target protein is known (structure-based virtual screening, SBVS). However, existing SBVS methods have two critical weaknesses: the tertiary structure of the target must be known, and their performance suffers greatly if the target protein undergoes structural changes when bound.
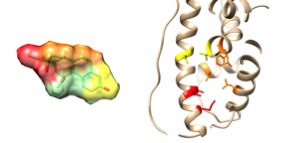
Professor Daisuke Kihara and Woong-Hee Shin, a postdoc in his group, have recently developed a new structure-based VS method, PL-PatchSurfer2. This method measures complementarity between a receptor pocket and a ligand by comparing local surface regions.
A unique strength of PL-PatchSurfer2 is its tolerance to structural variation in the target. By using a coarse-grained local surface descriptor representation, the method performs similarly on unbound or computationally modeled target structures and bound target structures: ~10% performance decrease compared to the more than 80% decrease suffered by existing methods. This level of performance enables the use of computationally modeled structures and low-resolution structures as targets, vastly increasing the number of potential target proteins for drug development, and opening a new path of drug discovery.
The work was recently published in J. Chemical Information and Modeling:
"PL-PatchSurfer2: Improved local surface matching-based virtual screening method that is tolerant to target and ligand structure variation". Woong-Hee Shin, Charles W. Christoffer, Jibo Wang, & Daisuke Kihara, J. Chem. Inf. Model [Epub ahead of print] (2016).