Purdue researchers successfully target 'Achilles' heel' of MERS virus
06-16-2015
A Purdue University-led team of researchers studying the Middle East Respiratory Syndrome, or MERS, have found molecules that shut down the activity of an essential enzyme in the virus and could lead the way to better treatments for those infected.
The virus is in the international spotlight again as South Korea faces the largest MERS outbreak outside the Middle East. More than 2,800 people have been quarantined during the outbreak. The World Health Organization reported 19 deaths and 154 confirmed cases in its most recent update.
The virus emerged in 2012 and was mostly confined to the Middle East until 2014 when cases were identified in the U.S., Britain, France and Italy. To date, 25 countries have reported cases, according to the WHO.
"The virus affects people differently and for many the symptoms are not life-threatening, but for others it can lead to severe respiratory distress," said Andrew Mesecar, Purdue's Walther Professor of Cancer Structural Biology and professor of biological sciences and chemistry who leads the research team. "It is a threat to public health we take very seriously and there currently is no treatment or vaccine. We continue to study the virus to improve our understanding of how it works and ways to prevent its spread."
Mesecar and Arun Ghosh, Purdue's Ian P. Rothwell Distinguished Professor of Chemistry and Medicinal Chemistry and Molecular Pharmacology, have been studying the virus and creating and testing molecular compounds that could lead to potential treatments since shortly after MERS was discovered.
The team identified molecules that inhibit an enzyme essential to MERS virus replication, and also discovered a characteristic of the enzyme that is very different from other coronaviruses, the family of viruses to which MERS-CoV belongs, Mesecar said.
"This enzyme is a prime target - an Achilles' heel of the virus - and we were excited to find an inhibitor that worked, but we were puzzled by the results," he said. "The behavior was very different from what our work with SARS and other related coronaviruses predicted. So, we investigated what was happening in order to put together the whole story. Now we have new, valuable information for the scientific community working on MERS."
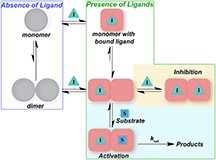
The team was targeting an enzyme within the MERS virus called 3C-like protease, without which the virus cannot create more viruses to further an infection. Once inside the cell, the virus creates a long strand of a large viral protein that must be cut at specific points to release individual proteins that serve various functions in building new virus particles. The 3C-like protease is responsible for making 11 of the necessary cuts for successful viral replication and without it the process shuts down, Mesecar said.
A single copy of the 3C-like protease must find and bond to another identical 3C-like protease "twin" in order to perform its function. Proteins that require bonding to a twin protein to perform their function are called dimers. All proteases in coronaviruses are dimers and most have a strong attraction to proteins of their identical type and bond very tightly to form the dimer.
Mesecar and his colleagues found that the MERS protease is unusual in that it does not have a strong attraction to its identical proteases and therefore does not readily form its dimer. This means an individual MERS 3C-like protease will remain single much longer and its dimer will break apart much more easily than the SARS protease or those of other coronaviruses, Mesecar said.
The team found that formation of the MERS protease dimer can be stimulated by the binding of a third molecule at a particular site on its surface to trigger the formation of a strong dimer. The particular site is where the protease would normally bond to the strand of protein it is meant to cut. When this bond is formed, the protease has an increased affinity for other 3C-like proteases and creates a stronger bond as it forms its dimer, he said.
This also was the site the team was targeting with an inhibitor molecule. By sending another molecule to attach to and block this key site, the protease would be unable to bind to the strand of viral protein and viral replication would be shut down.
However, there was a twist to what happened when the team began to add inhibitor molecules to interact with the protease. At low doses, the inhibitor increased the ability of a single MERS protease to find a twin, effectively activating the protease. Once the inhibitor bound to a single copy of the protease, it rapidly sought out a second identical protease to form a dimer. If the second protease had a vacant binding site, it was capable of binding to and cutting the strand of viral protein necessary for replication, Mesecar said.
As the team looked further into this unexpected result and increased the dose of the inhibitor, the scientists found that it would fill the target sites of all of the 3C-like proteases and its activity would be successfully blocked, he said.
"We were very surprised to see that this inhibitor molecule that could potentially shut down the virus may also have the potential to increase its activity," he said. "At low inhibitor concentrations we saw an increase in the protease's activity, but at high concentrations it was shut down completely. This makes it complicated as the work continues to turn this inhibitor into a viable treatment. We must be sure that all of the target molecules bind with the inhibitor."
A paper detailing their results was published in the Journal of Biological Chemistry on June 8 and is available online. The National Institutes of Health and the Walther Cancer Foundation funded the research.
In addition to Mesecar and Ghosh, Purdue co-authors of the paper include graduate students Sakshi Tomar, Heather L. Osswald and Prasanth R. Nyalapatla; postdoctoral researcher Sarah E. St. John; protein analysis research specialist Lake N. Paul; and undergraduate student Melanie L. Johnston. Mark R. Denison, an associate professor of microbiology and immunology at Vanderbilt University Medical Center, also is a co-author.
The team studied the interaction of the inhibitor molecule with 3C-like protease isolated from the MERS virus, but next plans to study the interaction of the inhibitor with a complete virus inside a cell, Mesecar said.
The team also will investigate other potential inhibitor molecules, he said.
"We captured the protease's atomic structure through this work, which provides the map to design potent new drugs to fight MERS," said Mesecar who also is deputy director of the Purdue University Center for Cancer Research.
Drug Discovery is one the key initiatives of Purdue Moves, a range of initiatives designed to broaden Purdue's global impact and enhance educational opportunities for students.
The MERS virus can spread from ill people to others through close contact, but has not been shown to spread within communities, according to the CDC. MERS causes severe respiratory illness, and symptoms include a fever, cough and shortness of breath.
Writer: Elizabeth K. Gardner, 765-494-2081, ekgardner@purdue.edu
Sources: Andrew Mesecar, 765-494-1924, amesecar@purdue.edu
Arun Ghosh, 765-494-5323, akghosh@purdue.edu
ABSTRACT
Ligand-induced dimerization of MERS coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals
Sakshi Tomar, Melanie L. Johnston, Sarah E. St. John, Heather L. Osswald, Prasanth R. Nyalapatla, Lake N. Paul, Arun K. Ghosh, Mark R. Denison and Andrew D. Mesecar
All coronaviruses, including the recently emerged Middle East respiratory syndrome coronavirus (MERS-CoV) from the ß-CoV subgroup, require the proteolytic activity of nsp5 protease (aka 3C-like protease, 3CLpro) during virus replication, making it a high-value target for the development of anti-coronavirus therapeutics. Kinetic studies indicate that in contrast to 3CLpro from other ß-CoV 2c members including HKU4 and KHU5, MERS-CoV 3CLpro is less efficient at processing a peptide substrate due to MERS-CoV 3CLpro being a weakly associated dimer. Conversely, HKU4, HKU5 and SARS-CoV 3CLpro enzymes are tightly associated dimers. AUC studies support that MERS-CoV 3CLpro is a weakly associated dimer (Kd ~52 µM) with a slow off-rate. Peptidomimetic inhibitors of MERS-CoV 3CLpro were synthesized and utilized in AUC experiments and demonstrate that MERS-CoV 3CLpro undergoes significant ligand-induced dimerization. Kinetic studies also revealed that designed reversible inhibitors act as activators at low compound concentration as a result of induced dimerization. Primary sequence comparisons and X-ray structural analyses of two MERS-CoV 3CLpro-inhibitor complexes, determined to 1.6 Å, reveal remarkable structural similarity of the dimer interface with 3CLpro from HKU4-CoV and KHU5-CoV. Despite this structural similarity, substantial differences in the dimerization ability suggest that long-range interactions by the non-conserved amino acids distant from the dimer interface may control MERS-CoV 3CLpro dimerization. Activation of MERS-CoV 3CLpro through ligand-induced dimerization appears to be unique within the genogroup 2c and may potentially increase the complexity in the development of MERS-CoV 3CLpro inhibitors as antiviral agents.