How to know which computational protein structure model is correct?
11-20-2014
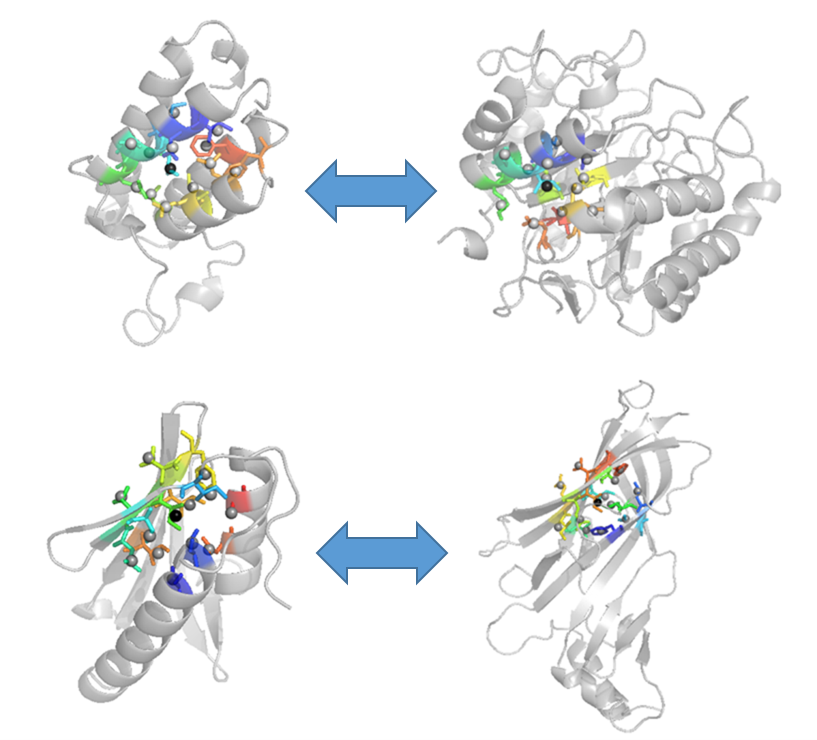
Computational protein structure modeling is routinely used for generating hypotheses and interpreting biochemical data of proteins. But out of many models you can generate using servers and software, which ones are more likely to be correct? Dr. Daisuke Kihara and Postdoctoral Research Associate Dr. Hyungrae Kim have developed a new procedure for finding near-native structure models.
Structural similarities and commonalities at various levels have been found in protein structures. Observed structural patterns are not only important for understanding the physical nature of the protein structures, but are also practically useful as a source of information for validating protein crystal structures as well as computationally predicted protein structure models.
In this study, published in the journal Proteins, the researchers developed a new representation of local amino acid environments in protein structures called the Side-chain Depth Environment (SDE). An SDE is determined by the coordinates and the depth of amino acids surrounding a particular amino acid residue. Similar SDEs are found in protein structures with globally different folds. Drs. Kihara and Kim developed a procedure called PRESCO (Protein Residue Environment SCOre), which compares model SDEs to a set of representative native protein structures to quantify similarity to native environments. When benchmarked on commonly used computational model datasets, PRESCO compared favorably with the other existing scoring functions in selecting native and near-native models.
Detecting local residue environment similarity for recognizing near-native structure models.
Kim H, Kihara D. Proteins. 2014 Jul 31. doi: 10.1002/prot.24658
Detecting local residue environment similarity for recognizing near-native structure models