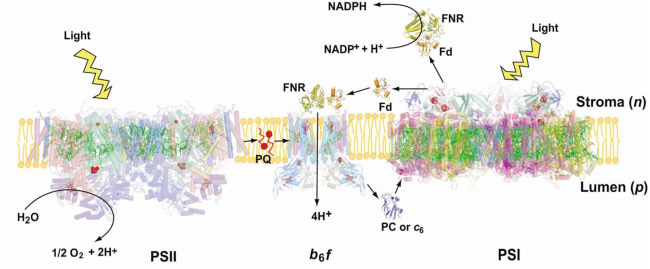
The cytochrome b6fcomplex in the electron transport chain of oxygenic photosynthesis. Photosynthetic electron transport is accomplished by integral membrane proteins: Photosystems I and II,and the cytochrome b6f complex. The cytochrome b6f complex occupies a central position in the sequence of photosynthetic electron transport carriers, oxidizing plastoquinol (PQH2) and providing the electron transfer connection between the two reaction center complexes, PS II and PS I, to which H+ transfer is coupled, thus contributing to the trans-membrane![]() . Electrons are transferred to PSI via plastocyanin or cyt c6 (Fig. 1). The proton-pumping cytochrome b6f complex has been shown by biochemical and mass spectroscopic analysis to be a dimer and to contain 8 tightly bound subunits/monomer (monomer MW = 108,500) in the cyanobacterium M. laminosus, and 9 in plant chloroplasts(Whitelegge et al ., 2002). The additional subunit in the complex isolated from spinach thylakoid membranes is ferredoxin: NADP- reductase [FNR] (Zhang et al, 2001), which is relevant to the mechanism of cyclic electron transfer linked to photosystem I. The b6f complex is in many respects analogous in function and structure to the cytochrome bcc1 complex from the mitochondrial respiratory chain and that of photosynthetic bacteria. Three of the polypeptide redox-active subunits in the b6fcomplex: cytochrome f, cytochrome b and the Rieske-type 2Fe-2S protein contain prosthetic groups. The two-electron oxidation of plastoquinol (PQH2) occur via a split pathway; one electron donated to the high potential [2Fe-2S] protein which is the electron donor to cytochrome f, the other to the pair of lower potential cytochrome b hemes.
. Electrons are transferred to PSI via plastocyanin or cyt c6 (Fig. 1). The proton-pumping cytochrome b6f complex has been shown by biochemical and mass spectroscopic analysis to be a dimer and to contain 8 tightly bound subunits/monomer (monomer MW = 108,500) in the cyanobacterium M. laminosus, and 9 in plant chloroplasts(Whitelegge et al ., 2002). The additional subunit in the complex isolated from spinach thylakoid membranes is ferredoxin: NADP- reductase [FNR] (Zhang et al, 2001), which is relevant to the mechanism of cyclic electron transfer linked to photosystem I. The b6f complex is in many respects analogous in function and structure to the cytochrome bcc1 complex from the mitochondrial respiratory chain and that of photosynthetic bacteria. Three of the polypeptide redox-active subunits in the b6fcomplex: cytochrome f, cytochrome b and the Rieske-type 2Fe-2S protein contain prosthetic groups. The two-electron oxidation of plastoquinol (PQH2) occur via a split pathway; one electron donated to the high potential [2Fe-2S] protein which is the electron donor to cytochrome f, the other to the pair of lower potential cytochrome b hemes.
Energy transducing proteins are prominent in the list of hetero-oligomeric integral membrane proteins, for which only about 2 dozen structures have been solved to a resolution of 3.0 Å. (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html). Studies on such complexes are of interest not only because of the insight they provide on their function, but for answers that they may provide to the problems associated with crystallization of integral membrane proteins, as summarized in (H. Michel, Crystallization of Membrane Proteins, 1991, 224 pp. CRC Press; S. Iwata, Membrane Protein Crystallography, 355 pp., Humana Press, 2004).
Crystallization and Structure of the b6f Complex
Crystallization of the cyanobacterial complex was impeded by proteolysis that rapid crystallization of the cyanobacterial complex, obtained from the thermophilic cyanobacterium, M. laminosus, was achieved (Fig. 2), and the race against proteolysis won (crystals obtained of b6fcomplex; below). Using a lipid augmentation strategy, as described in Zhang et al. (PNAS, 2003), the problem of optimization of lipid content was solved by adding 10 equivalents of synthetic lipid, DOPC, per monomer (n.b., the final DOPC concentration was 0.01%., diluted from a 2% stock solution containing 20% DMS0). Ribbon diagrams are shown (Fig. 3A,B) of the 217 kDa dimer, consisting of 8 subunits and 8 natural prosthetic groups (4 hemes, 1 [2Fe-2S] cluster, 1 chlorophyll a, one beta-carotene, and one plastoquinone per monomer (Zhang et al ., Science, 2003). A similar structure was solved from C. reinhardtii by Stroebel et al . (Nature, 2003).
 Fig. 2. Crystals of the b6fcomplex from M. laminosus grown in the presence of DOPC lipid.
Fig. 2. Crystals of the b6fcomplex from M. laminosus grown in the presence of DOPC lipid.
The b6f complex is a 220 kDa symmetric dimer (Figs. 3A, B). Ribbon diagrams are shown of the dimeric b6f complex containing 8 subunits, 13 trans-membrane helices and 7 prosthetic groups (4 hemes, 1 [2Fe-2S] cluster, 1 chlorophyll a and 1 ?-carotene) (Figs. 3A, B). One of the 4 hemes is the unique heme cn that is covalently bound to a Cys residue on the n-side of the cyt b A helix. The core of the b6f complex, consisting of the cyt b6 and subunit IV polypeptides is shown in blue in the view parallel to the membrane plane (Fig. 3A), and in green in the orthogonal view (Fig. 3B). The cyt b core is homologous between bc, and b6f complexes with respect to the distribution of hydrophobicity and the His residues that ligate the hemes. Away from the central core parallel or perpendicular to the membrane plane, the b6fand bc1 structures are less similar. The rubredoxin-like core of the ISP protruding into the p-side aqueous phase (Fig. 3A) has a similar fold in the 2 complexes, but the ISP domain distal to the membrane is different. Except for the 5 residue heme binding sequence Cys-X-Y-Cys-His, cyt f is completely unlike cyt c1, in primary sequence, secondary structure, and axial heme ligation. The cavity between the 2 monomers defines the quinone exchange cavity, the space in which quinone(ol), transferred from the membrane bilayer, is confined as it carries out n- to p-side H+ transfer and electron transport. The p- and n-side surface potentials are negative (– 5.4 kT, Fig. 3C, left; cyt f and the ISP are acidic proteins) and positive (+ 4.6 kT, Fig. 3C, right). The 2 b-hemes (magenta, Fig. 3B) bridge the B and D trans-membrane helices of the cyt b polypeptide. The Chl phytyl chain is wrapped around the F -helix, emerging through a portal to the quinone exchange cavity.
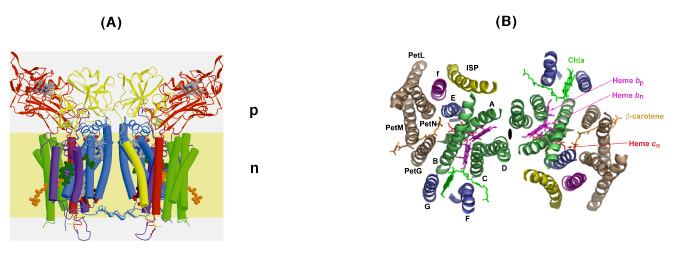
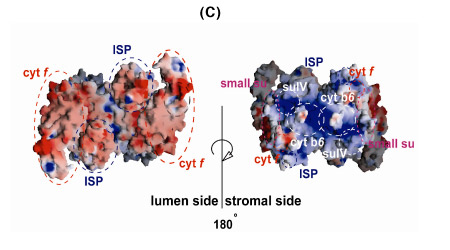
Fig. 3. Views (A) parallel and (B) normal to the membrane plane of dimeric cyt b6fcomplex: MW = 220,000; 13 TM helices per monomer, 8 subunits, 7 prosthetic groups (4 hemes, including heme cn, 1 [2Fe-2S] cluster, 1 Chl a, 1 ?-carotene); 30 Å high x 25 Å wide (n-side) x 15 Å deep inter-monomer cavity that connects the QpH2 oxidation site in 1 monomer with the Qn reduction site in the other. 4 TM helices of cyt b6, green; 3 of suIV, blue; cyt f, purple; ISP, yellow; 4 small hydrophobic subunits, petG, L, M, N, gray-brown, define a structure at the periphery of each monomer through which the ?-carotene penetrates, one iodinylene ring contacting subunit IV. (C) Asymmetry in p and n- side surface potential, -5.4 kT and +4.6kT, respectively. The positive surface potential on the n-side results mostly from cyt b basic residues Arg207 and Lys208.
Similarities and differences in function between bc1 and b6f complexes. There is universal agreement that both bc1 and b6f complexes operate to transfer H+ across the complex by oxidizing a lipophilic quinol (Table 1, formula [1]) through a high potential electron transport chain, containing the [2Fe-2S] ISP, cyt f , PC or cyt c6 and the PSI reaction center. It is often argued that the b6f complex functions identically to the bcc1 complex in an identical Q cycle function. However, the presence in the b6fcomplex of the heme cn and the ferredoxin (Fd) -dependent cyclic electron transport pathway (Fig. 1) that can provide additional ATP, suggests that the Q cycle may not be obligatory in the b6f complex. An experimental difficulty in analysis of function of the b6fcomplex is that, unlike the bc1 complex, the two b hemes of the b6fcomplex are not readily spectrophotometrically distinguishable, and there is no high affinity quinone analogue inhibitor like antimycin A to thoroughly block electron transfer on the n-side of the complex.
The following reactions (Table 1) define the electron/proton transfer pathways in a ‘modified Q cycle’ summarize electron and proton transfer reactions in the b6f complex. Reactions [1-4] are unchanged from a conventional Q cycle formulation in which the n-side quinone is reduced through a low potential chain containing hemes bp and bn, b-type hemes on the p- and n-sides of the complex. However, formulae [5-8] take into account the ideas that hemes bn –cn form a 2 electron transfer system (Cramer et al., 2006b; Zatsman et al., 2006; Yamashita et al., 2007) that can be reduced not only by p-side plasto-semiquinone and the low potential heme bp – heme bn pathway, but also by ferredoxin/FNR via the PSI-linked cyclic electron transport pathway (Figs. 1, 4).
Table 1. Summary of Electron/Proton Transfer Reactions in the Cytochrome b6f Complex
| PQH2 + [2Fe-2S (ox)] → PQ.- + [2Fe-2S (red)] + 2H+ [p-side aqueous phase] | [1] |
| FeS (red) + cyt f (ox) → FeS (ox) + cyt f (red) [high potential chain; n. b., structure indicates ˜ 28 Å for center-center distance of transfer; subsequent steps in the chain are cyt f → PC or cyt c6 → P700] |
[2] |
| PQ |
[3] |
| PQ |
[3a] |
| 2 heme bp(r) + hemes bn(o)/cn(o) → 2bp(o) + bn(r)/cn(r) [two turnovers of p-side PQH2 oxidation; trans-membrane electron transfer to bncn,/ complex] |
[4] |
| bn(r)/cn(r) + 2H+ + PQ → bn(o)/cn(o) + PQ.H2 [n-side 2e- reduction of PQ, using the heme bncn 2 electron gate]. |
[5] |
| 2 Fd (red) + FNR (ox) → 2 Fd (ox) + FNR (red) | [6] |
| FNR (red) + heme bn (ox)/cn (ox) → FNR (ox) + heme bn(red)/cn (red) | [7] |
| bn (red)/ cn (red) + 2H+ + PQ → 2bn(ox)/cn (ox) + PQH2 | [8] |
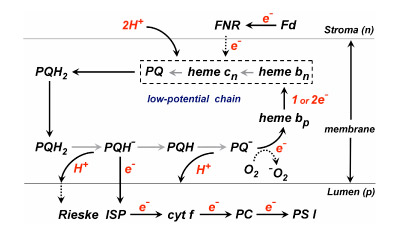
Fig. 4. Model for the pathway of electron and H+ transfer in the cyt b6f complex based on the “Q cycle” model developed for the bc1 complex. Major differences in the b6f complex are: (i) the presence of the additional heme, cn, coupled closely and tightly to heme bn; (ii) electron input into b6f through ferredoxin and perhaps FNR. Properties of heme cn are discussed below. The order of electron transfer is believed to be bn to cn, or concerted 2 electron transfer from coupled hemes bn/cn to reduce PQ to PQH2.
The Rieske Iron-Sulfur Protein ISP). It has been shown in the in the bc1 complex that the Rieske high potential [2Fe-2S] cluster can accomplish long distance electron transfer, from a membrane proximal site where it is reduced by the bound quinol, to a site near cyt c1 where it is oxidized, by undergoing a rotation-translation of approximately 60º and 17 Å, pivoting around a flexible loop that connects the soluble domain in one monomer with a trans-membrane helix that spans the membrane in the other monomer. The different position of the cyt f heme, on top of the elongate cyt f implies that that electron transfer from the [2Fe-2S] cluster will go through the cyt f apoprotein and that the ISP motion will be of smaller amplitude. We have shown that ISP electron transfer function in the b6f complex is less sensitive to mutations that decrease the flexibility of the ISP ‘flexible’ loop.
NDH-1. As in the bc1 complex, the quinone(ol) pool (PQ in Fig. 2) that serves as the source and sink of the electron and proton flux through the b6f complex can also be reduced by an NADH dehydrogenase (a 15 subunit NDH in the photosynthetic system). The analogous complex I is the only major electron transport complex in the respiratory chain that has not been solved although a structure has been obtained of the extrinsic domain. We have isolated an NDH preparation from the thermophilic cyanobacterium, Thermosynechococcus elongatus, supplied by E–M. Aro, and with J. Whitelegge have identified 12 of its subunits by mass spectrometry.
Evolutionary origin of heme cn. The absence of heme cn in the bc1 complex implies that a plausible function of this heme in the b6f complex is in the pathway of photosystem I-linked cyclic electron transport (Fig. 1). However, the evolutionary origin of heme cn is close to the firmicute or gram-positive branch of the evolutionary tree, where it would obviously have a non-photosynthetic function, possibly in a 2 electron reduction of plastoquinone that that would avoid superoxide formation (Table 1; reaction 5). In addition, the gene sequence motifs coding for both hemes b and cn are seen not only in cyanobacteria, but also in the non-photosynthetic firmicutes that include gram-positive bacteria such as B. subtilis . The presence of the conserved residues Cys35 that serves as a ligand for heme cn, and His86, 100, 187, and 202, which serve as ligands for hemes bp and bn, can be noted. The presence of a complex in the gram-positive Bacillus subtilis and in B. Stereothermophilus, which resembles the cytochrome b6f complex, and the demonstration of a covalently (cysteine-) bound c-type heme in the QcrB (cyt b) subunit, implies a metabolic role for heme cn that is presently unknown in the primitive non-photosynthetic micro-organisms. The cyt bc complex has been proposed to be essential for maintaining the redox poise of the lower potential (Em7 ≌ - 60 mV) menaquinone redox couple that also functions in as an electron and proton acceptor in the succinate:quinone oxidoreductase of B. subtilis. It is of interest that the midpoint (Em7) of the menaquinone redox couple (MQ/MQH2) is ~150 mV more negative than that of ubiquinone or plastoquinone.
Summary of cyanobacterial b6f crystal structure data: Crystal structures have been obtained and deposited of the native b6f complex (pdb accession: 2E74), and the complex in the presence of the quinone analogue inhibitors DBMIB (2D2C), NQNO (2E75), and tridecyl-stigmatelin (2E76).
Structure of b6f complex in the native state. The native structure of cyanobacterial b6f was solved initially at 3.4 Å, with refinement parameters Rcryst = 0.256, Rfree = 0.336. The improved refinement parameters for the native structure solved to 3.0 Å in the presence of divalent (Cd2+) cations are: Rcryst = 0.222, Rfree = 0.268. The increase in resolution and quality of refinement parameters is significant because (i) there is no other native structure of the b6f complex; (ii) assignment of inhibitor binding sites can be made with a much greater degree of certainty; (iii) it has not yet been possible to obtain a native structure of comparable resolution of the cytochrome bc1 complex, because of disorder in the Rieske ISP. Thus, most bc1 structures have been obtained in the presence of a p-side inhibitor such as stigmatellin, which stabilizes the Rieske ISP and locks it into a membrane-proximal position.
Cd2+ binding sites. Although binding of the Cd2+ cation allowed a higher resolution native structure to be obtained, this ion has no special role in the function of the b6f complex. A search was made for divalent ions to stabilize the complex. Cu2+ and Zn2+, the more obvious divalent ions that are known to bind to the bc1 complex, were tried, but it was not possible to obtain well diffracting crystals. In the improved native structure, the binding sites of Cd2+ were determined from the anomalous difference map for Cd2+. The two divalent Cd2+ cation sites are close to the p-side membrane interface of the complex in each monomer, and bridge two p-side basic residues of cyt f (His 143 and Lys146) with a carboxylate of cyt b6 (Glu75) and subunit IV (Asp58), and 1-2 H2O or Cl- ions [Figs. 5A, B].
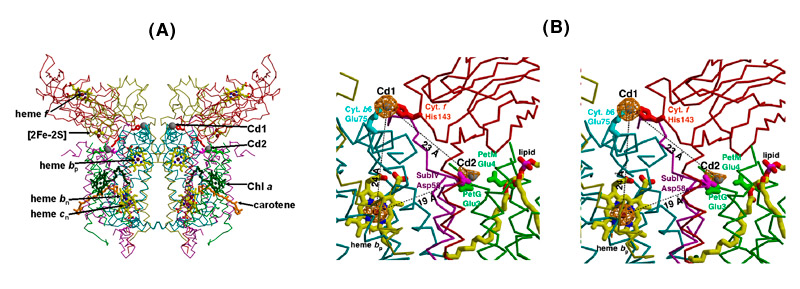
Fig. 5. Two binding sites of the divalent cation, Cd2+, in the b6f complex,(A) seen in the perspective of the dimeric structure; (B) magnified view in stereo; sites determined by an anomalous difference map at 3.6 Å, bridging His143 of cyt f to Glu75 of cyt b6 (Cd site I) and two Glu and 1 Asp residues (Cd site 2); Cd atoms, gray balls; cyt b6, cyan; suIV, purple; cyt f, red; ISP, yellow [pdb accession, 2E74].
Structure of b6f complex in the presence of quinone analogues; cyt cn as a quinone binding site
Quinone analogue inhibitors: Tridecyl-stigmatellin (TDS) and 2-n-nonyl-4-hydroxquinoline-N-oxide (NQNO). Three results of interest were obtained: TDS was found to bind on both sides of the complex. (i) On the p-side, the TDS chromone ring was found to reside within an H-bond distance of the His129 ligand to the membrane-proximal Fe of the [2Fe-2S] cluster. This modified a previous result from these studies in which TDS was oriented “chromone ring-out,” the latter result a consequence of (a) the lipid addition that was necessary for crystallization being done prior to addition of TDS, which prevented proper binding of TDS, and (b) the absence at that time of a native structure of sufficient quality to allow comparison with a control electron density. (ii) TDS, which as a stigmatellin derivative, is a classical p-side inhibitor, was also found to bind on the n-side of the complex, as an apparent ligand to the free axial position of heme cn. Heme cn is shown in the absence and presence of TDS in Figs. 6A, B. The R-factors for the refinement of the TDS structure are Rcryst = 0. 201 and Rfree = 0.258. (iii) The classical n-side inhibitor, NQNO, was also found to bind as a ligand to heme cn (Fig. 6C).
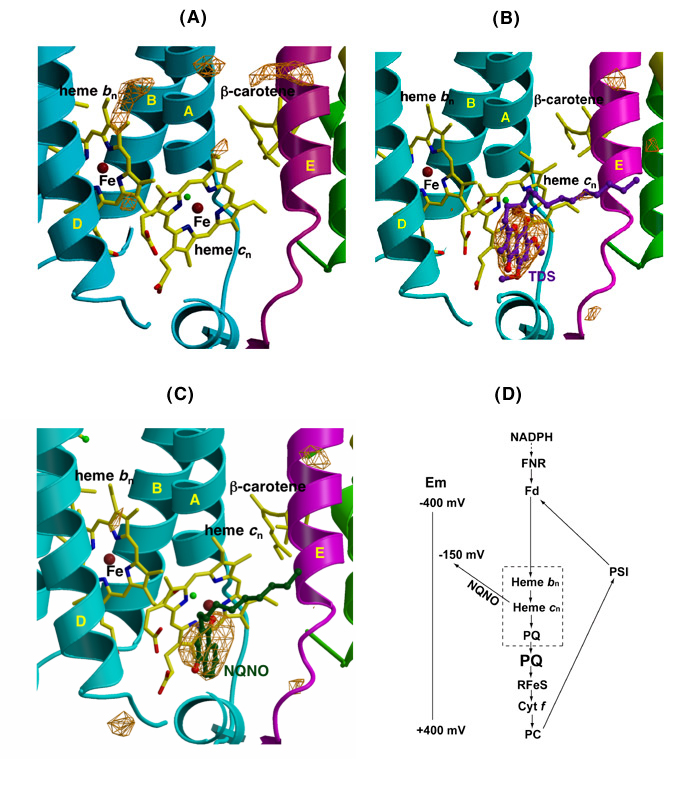
Fig. 6. Ribbon diagrams from Fo –Fc difference maps of the n-side of the b6f complex (A) in the absence of inhibitors, (B) in the presence of TDS (pdb accession, 2E76), and NQNO (C) (pdb, 2E75); the two inhibitors bind as ligands of heme cn; (D) model for the electron transport chain in the b6f complex, showing the n-side electron and proton transfer complex formed by quinone and heme cn (in dashed box), the reduction of the n-side complex by reduced ferredoxin/FNR, and the shift in redox potential of heme cn caused by bound NQNO, which is proposed to be the basis for inhibition by NQNO.
The inhibitory action of NQNO on the b6f complex has some resemblance to that by antimycin A in the cytochrome bc1 complex. Thus, its presence is known to cause an increase in the amplitude of cyt b reduction by a light flash.
Heme cn as the n-side quinone binding site. From the binding of the two quinone analogue inhibitors to the open axial position of heme cn, it is inferred that this is the physiological binding site for the n-side plastoquinone. The complex formed by heme bn – cn –PQ, shown as a dashed box in Figs. 6, 6D, is the n-side pathway for transferring electrons and protons to the PQ pool, and is functionally analogous to the n-side bn – UQ segment in the cyt bc1 complex. The electron transport chain described in Fig. _D also emphasizes (i) that the mechanism of inhibition by NQNO is the --200 mV shift in midpoint potential of heme cn caused by bound NQNO, and (ii) the ferredoxin-dependent cyclic electron transport pathway, summarizing the result that reduction of hemes bn - cn by NADPH requires both ferredoxin and FNR.
p-side inhibitor, 2,5-dimethyl-6-isopropyl benzoquinone (DBMIB). Using the M. laminosus b6f complex, structure data was obtained for the high affinity binding site of the quinone analogue inhibitor,DBMIB, and on the ability of the DBMIB to move from this site to the [2Fe-2S] cluster after illumination. The high affinity DBMIB binding site, 19 Å from the [2Fe-2S] cluster (shown below in Fig. 7), is the most extremely positioned p-side inhibitor binding site in that has been determined.
Parameters of the Structure. The Subunit masses, pI values, midpoint potentials and intra-complex distances are summarized (Tables 3,4)
|
Table 3. Subunit masses, pI values, and midpoint redox potentials at pH 7 (Em7) of cyt b6f subunits of M. laminosus
*suIV, 4 small subunits: no bound redox prosthetic group |
Table 4. Intra and inter-monomer distances between prosthetic groups in the M. laminosus b6f complex.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Trans-membrane traffic of quinones, quinone-containing drugs. Energy–transducing trans-membrane traffic is not limited to electrons and protons, but also includes hydrophobic quinones that move across these protein complexes not by a simple “flip-flop” mechanism that was an early concept, but by a ‘guided’ diffusion through a labyrinthine pathway. An understanding of the pathways, and structure requirements for transfer of the quinone(ol) across across the b6f and bc1 protein complexes is relevant to the pathways and mechanisms utilized by quinones and quinone-analogue anti-cancer drugs (e. g., adriamycin, mitomycin C, lapachol, and vitamins (e. g., vitamin K).
Passage of the lipophilic quinone across the b6f complex. The pathway and “guided diffusion” mechanism by which the plastoquinone with its lipophilic long tail containing 9 isoprenoid groups and 45 carbon atoms, crosses the complex, passing across the lipid-filled 30 Å x 25 Å x 15 Å quinone exchange cavity between its p-side oxidation site and n-side reduction site is not understood. However, the pathway of Q/QH2 across the complex is marked by the binding sites of different quinone analogue inhibitors. The binding sites of 3 such analogues, tridecyl-stigmatellin (TDS, binds on the p-side of the quinone exchange cavity), DBMIB (p-side), and NQNO (n-side), span the complex from the p- to the n-side, and thereby mark the path of movement of PQ/PQH2 (Fig. 7).
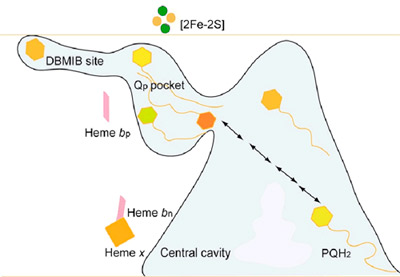
Fig. 7. Representation of the labyrinthine pathway of plastoquinone between p-side oxidation and n-side reduction sites. Prominent features are the inter-monomer quinone exchange cavity, the narrow 11 x 12 Å portal from the cavity to the p-side quinone binding pocket, and the extreme peripheral location of the high affinity ‘dark’ site of DBMIB.
The portal connecting the quinone exhange cavity and the p-side quinone binding niche. The physiological plastoquinone and the p-side quinone analogiue inhibitors (e. g., TDS) are inserted from the inter-monomer quinone exchange cavity through an ~ 11 Å x 12 Å portal in the “roof” of the cavity, to reach the [2Fe-2S] cluster in the other monomer at the p-side interface of the complex (Fig. 8). The phytyl chain of the bound chlorophyll a, whose ring is inserted between the F and G helices of subunit IV (light green prosthetic group in Figs. 9A, B), is wrapped around the G helix (subunit IV) and is also inserted through the portal. Mutagenesis experiments were conducted to determine the identity of the residue(s) responsible for passage of stigmatellin through the portal. The identity of residue 81, which is Leu in wild type cyanobacteria and Phe in higher plants and algae, had a large effect on the sensitivity of electron transport to TDS.
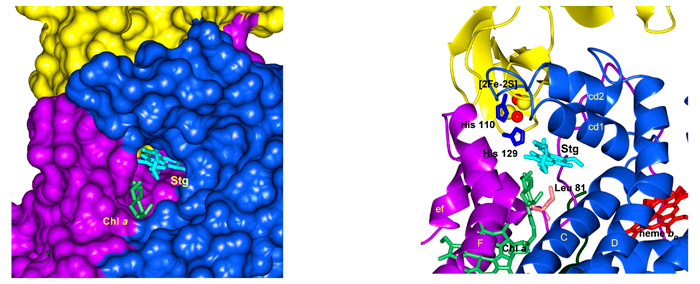
Fig. 8. (A) Representation in PYMOL of the 11 Å x 12 Å portal on the roof (p-side) of the inter-monomer quinone exchange cavity, showing the tail of TDS (magenta) emerging from the portal into the cavity. Chl head group in green is positioned between the F and G helices and the Chl phytyl chain is wrapped around the F helix. (B) Stick and ribbon diagram of stigmatellin emerging from portal into cavity; Residue Leu81 that has a large effect on entry into the portal ), and the imidazole ring of His 129, which forms an H-bond with O-4 of the stigmatellin ring, are shown.
Structure-function of heme cn. Given the absence of any amino acid side chains as axial ligands in both the cyanobacterial and green algal structures, heme cn, a heme on the n-side of the complex that is covalently bound to the cyt b subunit by one cysteine (Cys35, Fig. 9) is a unique protein-bound heme. Using the ‘i’ –‘o’ (inside-outside) notation from the mitochondrial bc1 complex, cn is sometimes called cytochrome ci. although ‘inside/outside’ is reversed in membranes of oxygenic photosynthesis compared to those of mitochondrial respiration. membranes. Structurally, heme cn is very close to the n-side heme bn so that the only ligand to the heme Fe of cn is a water molecule that bridges 3.5 Å from the heme cn Fe to a propionate O atom of the neighboring heme bn (Fig. 10).
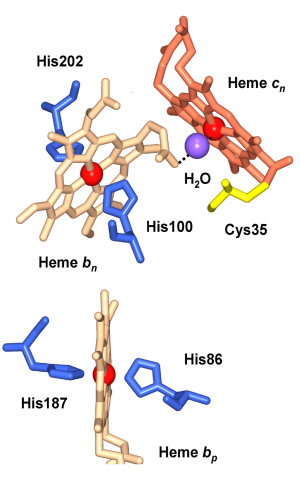
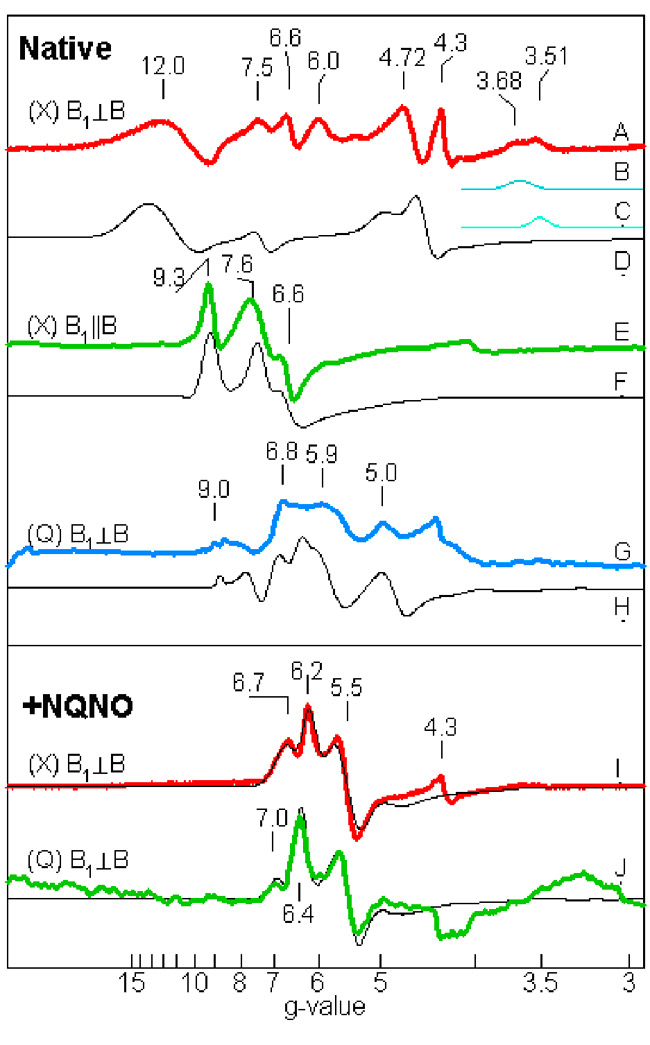
Fig. 9 (left). Hemes bn and cn near the electrochemically negative side of the b6f complex showing the proximity of the two hemes and their orthogonal orientation. The ligation of the cn Fe by a hydroxyl or H2O molecule is shown, as are the histidine ligands to heme bn, Cys35 that forms the covalent linkage with heme cn, and a neighboring residue, Phe40, which is in a conserved region.
Fig. 10 (right). Multi-frequency EPR spectra of exchange-coupled spin system of cyt b6f complex (0.1 mM cyt f, pH7.5). Spectra: A- H, native; I, J, with NQNO, A-F: X-band; G, H, J: Q band; E,F: parallel mode. (Zatsman, et. Al., 2006); (Measurements made in lab of M. Hendrich)
The initial characterization of heme cn by EPR showed it to have a high spin nature. The proximity of the two hemes, which implies very rapid electron transfer and exchange, raised the question of whether this property is reflected in the EPR spectra. Recent EPR experiments conducted with M. Hendrich (Carnegie-Mellon University), which used parallel mode detection and measurements at low values of the magnetic field able to probe for high ‘g’ values that would result from coupling of the hemes did, indeed, show elevated g values, up to g = 12. (Fig.10). Together with signal detection in parallel mode, with the radio frequency magnetic field vector parallel to that of the static strong orienting magnetic field (a geometry that cannot yield a signal from a single high spin heme), and the close (3.5 Å) structural proximity (Fig. 10), implies a strong coupling between the two hemes. The biological implication is the possibility of a 2 electron transfer to its electron acceptor (Zatsman et al., 2006), as described in the electron transport scheme of Fig. 4. From the observation of NQNO as a distal-side ligand to the Fe of heme cn (Fig. 6C), and the appearance of electron density associated with plastoquinone in the absence of NQNO, it was inferred that plastoquinone is the immediate electron acceptor of the heme bn -heme cn couple (Fig. 6D).
High resolution structure and function studies on cytochrome f and the Rieske [2Fe-2S] proteins
Cytochrome f is unique among c-type cytochromes because:
- it is mostly b-strand (figure, below left), an elongate 75 Å structure consisting of a small and large domain.
- it has a buried chain of 5 water molecules (red balls for O atoms) spanning 11 Å (figure, below left).
- one of the axial heme ligands is the alpha-amino group of the N-terminal amino acid residue (figure, below center).
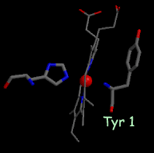
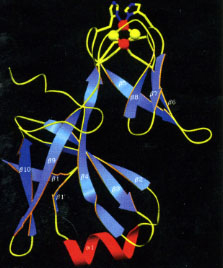
High-resolution structures of the soluble domain of cytochrome f from a plant (turnip), a cyanobacterium (P. laminosum) and a green alga (C. reinhardtii) are available from the protein data bank. (far right)1.85 Å structure of the Rieske [2Fe-2S] protein from spinach thylakoids (Carrell et al., 1997).”
Interaction of cytochrome f with its electron acceptor, plastocyanin
Complementary surface charges near the redox centers and ionic-strength dependence of rates of oxidation of cytochrome f by plastocyanin in solution indicate the presence of electrostatic interactions between the redox partners. However, neutralization of the basic charges on cytochrome f through site-directed mutations did not affect reaction rates in vivo implying that in vivo, cyt f-plastocyanin electrostatic interactions are not required.
Structure-function studies on the Rieske 2Fe-2S protein
The Rieske protein consists of a soluble 140-150 residue domain in a beta-sheet conformation (Figure, above, right-hand panel) anchored to the membrane by one trans-membrane helix that is obliquely oriented so that it is "domain-swapped" and crosses over from one monomer to the other (see yellow helix in Figure 3A describing b6f dimer, ), so that the total molecular weight of the Rieske protein is approximately 20,000. The structure of the soluble domain of the Rieske protein was determined using crystals of the soluble domain (Carrell et al., Structure, 1997).
Electron-transfer via large amplitude rotation-translation. The first events in charge transfer from the lipophilic quinol bound near (H-bound distance to the quinone ring) are transfer of a proton and electron transfer to the Rieske 2Fe-2S protein bound in a membrane proximal position. Subsequent electron transfer through the high potential chain to the membrane bound c-type cytochrome was shown in the mitochondrial bc1 complex to involve a large amplitude rotation (approx. 60°) - translation (approx. 16 Å) to bridge the very large (25 Å) distance between the FeS cluster and the heme of cytochrome c1 (Zhang et al., Nature, 1998). The flexible hinge region of the Rieske protein that connects the soluble and membrane-bound domains serves as the pivot of the rotation-translation. The function of the bc1 complex was shown to be very sensitive to mutational change in the hinge region. The distance of the 2Fe-2S cluster to the cytochrome f heme in the b6f complex is similar, and it is assumed that electron transfer between these centers also requires a large scale movement. This is consistent with a systematic effect of medium viscosity of the rate of cytochrome f reduction. However, function of the cytochrome b6f complex was much less sensitive to similar mutational changes in its glycine - rich hinge region (Yan and Cramer, JBC, 2003), suggesting that the amplitude of the ET-coupled rotation-translation of the Rieske soluble domain is smaller than in the bc1 complex as discussed in Kurisu et al., Science (2003).