Project
BackgroundJustification
Proposed Experiments
Distribution of Project Resources and Information, and Intellectual Property Issues
Outreach
Background
A. The Agrobacterium T-complex
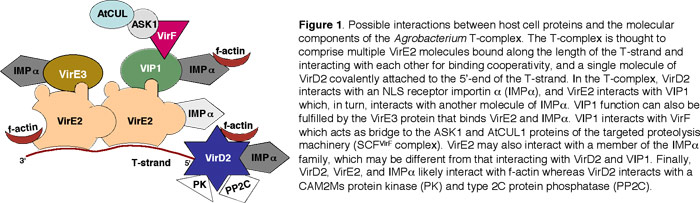
Agrobacterium tumefaciens elicits neoplastic growths on many plant species (15). Although plants represent the natural hosts for Agrobacterium, this microorganism can also transform a wide range of other eukaryotic species, from fungi (16, 62) to human cells (48). This genetic transformation is achieved by transporting a single-stranded copy (T-strand) of the bacterial transferred DNA region (T-DNA region) from the tumor-inducing (Ti) plasmid into the plant cell nucleus, followed by integration into the host genome (reviewed in 29, 30, 81). In Agrobacterium, the T-region is delimited by two 25-bp direct repeats, the T-DNA borders, that are cleaved by the bacterial VirD2 endonuclease to produce a transferable T-strand molecule (86). Whereas only the wild-type T-DNA carries tumor-inducing genes, any DNA placed between the T-DNA borders will be transported into the plant cell nucleus (reviewed in 30, 90). This lack of sequence specificity implies that the T-DNA molecule itself does not contain specific signals for cytoplasmic trafficking, nuclear import, and integration. Instead, these processes are likely mediated by two Agrobacterium Virulence (Vir) proteins, VirD2 and VirE2, that are thought to associate directly with the T-strand, forming a transport (T) complex (97). In the current model of the T-complex, one molecule of VirD2 is covalently attached to the 5'-end of the T-strand whereas VirE2, a single-stranded (ss)DNA binding protein, cooperatively coats the rest of the ssDNA molecule (29,30,38,81,96,97) (Figure 1).
The proposed T-complex of wild-type Agrobacterium can be quite large, reaching up to 9x104 kDa and carrying approximately 1,200 molecules of VirE2 (10). The 12.6-nm calculated diameter of the T-complex (10) exceeds the diffusion limit of the nuclear pore (9 nm, reviewed in 24) but is compatible with the 23-nm size exclusion limit of the active nuclear import (21). T-complex nuclear uptake is actively mediated by its cognate VirD2 and VirE2 proteins ( 94), which have been shown to be targeted to the host cell nucleus (11, 12, 39, 47, 55, 69, 80). Furthermore, VirE2 alone is sufficient to transport ssDNA into the nucleus of the plant cell (95), and may be responsible for T-complex nuclear import if the NLS sequences are deleted from VirD2 (28).
B. Host cell proteins that likely interact with the T-complex
In plant cells, the T-complex likely interacts with cellular factors that mediate cytoplasmic trafficking, nuclear import, and T-DNA integration (Figure 1). Several plant proteins have been identified that, at least in yeast, interact with VirD2, VirE2, and with the bacterial host range factor VirF (4, 5, 18, 29, 72, 80). For example, several (but not all) tested importin ? proteins interact with VirD2 (4, 5; S. Bhattacharjee and SBG, unpublished), and mutation of importin ?-7 (but not other importin ? genes) results in loss of transformation proficiency of Arabidopsis (rat, resistance to Agrobacterium transformation, phenotype). Importin ? also interacts with VirE3 (VC, unpublished). Other Arabidopsis proteins that interact in yeast with Vir proteins include VIP1 with VirE2 (82), a type 2C protein phosphatase (Y. Tao and SBG, submitted) and a CAK2Ms protein kinase (4) with VirD2, F-actin with VirD2 and VirE2 (P. Rao and SBG, unpublished), and ASK1 (the plant homologue of the yeast Skp1p) with VirF (72; VC, unpublished). Because mutation of numerous chromatin protein genes results in the rat phenotype (92; Y. Crane and SBG, unpublished), it is also likely that at least some of these proteins (including histone variants, acetyltransferases, deacetylases, and chromatin remodeling factors) interact with Vir proteins found in the T-complex.
What has become increasingly clear is that the T-complex likely interacts with a large number of host factors that mediate cytoplasmic trafficking (F-actin), nuclear targeting (VIP1, protein phosphatases, importins, and possibly transportins), T-strand "uncoating" in the nucleus through targeted proteolysis (F-box proteins), and T-DNA integration (histones, other non-histone chromatin proteins, and a protein kinase). Thus, Agrobacterium T-complex trafficking through the plant cell is an excellent and unique model system to study a large number of important and diverse plant cell biology processes.
In this proposal, we shall develop a novel technology, termed Bimolecular Fluorescence Complementation (BMFC), to study protein-protein interactions in plants. The vectors that we shall develop, and the cDNAs that we shall express from these vectors, will serve a large segment of the plant cell biology community. In addition, we shall generate cDNA libraries in these novel vectors that can be used as "preys" to investigate protein-protein interactions in planta with "bait" proteins of choice. As a proof of concept, we shall investigate interactions of Vir proteins of the Agrobacterium T-complex with Arabidopsis protein families involved in such general cellular pathways for processing of nucleoprotein complexes as cytoplasmic trafficking (cytoskeletal proteins), nuclear targeting (importins, transportins, and VIP1), targeted proteolysis (F-box proteins), and T-DNA integration (chromatin proteins).
Justification
This proposal meets the goals of the 2010 project in two ways: 1) We shall develop new technology (BMFC) that will serve the needs both of the Arabidopsis and entire plant community. Currently, there does not exist the equivalent of a "plant two-hybrid" system to investigate protein-protein interactions in planta. This project will provide such a system. 2) This project will provide an innovative technology to investigate networks of plant genes involved in Agrobacterium-mediated transformation. The proteins encoded by these genes are involved in a vast array of important cell biology functions, and knowledge of the interactions of specific protein pairs will be essential both for dissecting these functions and for understanding and improving Agrobacterium-mediated transformation.
Proposed Experiments
A. Bimolecular Fluorescence Complementation (BMFC) to investigate protein interactions in planta
We shall utilize bimolecular fluorescence complementation (BMFC) to investigate
protein-protein interactions in planta. Hu et al. (40) first described this
system for use with animal cells, and Atmakuri et al. (2) recently utilized
this approach to investigate the interaction of VirE2 and VirD4 proteins in
Agrobacterium cells. (Please note that VirD4 is a membrane-associated
protein, so BMFC can work for membrane proteins). In the BMFC system,
a GFP or YFP molecule is split into two portions (for simplicity, hereafter
termed "halves" although they are not equal in size, see below for details),
neither of which fluoresces on its own. However, when these two YFP portions
are brought together as fusions with interacting proteins, fluorescence is
restored. The BMFC design serves as a novel in planta "two-hybrid" system
with five major advantages over other approaches for studies of protein-protein
interactions:
- The interaction is detected in planta, circumventing the major flaw of most other protein interactions systems, such as the yeast two-hybrid assay, of extrapolating the interaction data from a heterologous system to plants.
- Individually, the BMFC-tagged proteins produce no fluorescence at all, eliminating background signal that often complicates such interaction assays as co-immunoprecipitation (co-IP).
- The interaction is detected directly by appearance of the fluorescent signal, avoiding labor-intensive and time consuming colorimetric staining, selection assays, BRET/FRET, or co-IP.
- This direct and easy detection allows its use for high-throughput experiments essential in most genomics projects.
- The fluorescent signal indicating protein-protein interaction also identifies the subcellular localization of the interacting proteins (39).
Note that BMFC was originally developed by Dr. Changdeng Hu for use in animal
systems (39). Dr. Hu is presently an Assistant Professor at Purdue University,
and has agreed to advise us on the technical aspects of this project (see attached
letter).
1. Generation of Constructions to use for BMFC
Because the exact structure of interacting proteins is usually not known, to minimize the interference of the half-YFP tag with the protein-protein interaction, and to maximize the probability that the two halves of the YFP molecule will be close enough to generate the fluorescent signal, we have designed vectors to express either N-terminal or C-terminal portions of YFP (YFP-N and YFP-C, respectively) that can be fused to either the N-terminus or the C-terminus of proteins to be investigated. Thus, for any given protein pair, there can be up to eight constructions (YFP-N at the N-terminus of each protein, YFP-N at the C-terminus of each protein, YFP-C at the C-terminus of each protein, and YFP-C at the N-terminus of each protein) and four corresponding combinations of proteins to test. Our constructions will also incorporate an amino acid "linker" to "distance" the protein of interest from the half-YFP tag (see below); such linkers are known to act as flexible tethers that minimize potential folding interference between two fused proteins (20). One of the co-PIs (VC) has tested the DNA sequence encoding the YFP protein that we shall use to assure that it expresses well in plants and does not contain the cryptic splice sites and polyadenylation signal sequences recognized by Arabidopsis (35).
We shall first clone the cDNAs encoding the proteins of interest into a Gateway (Invitrogen) entry vector. We shall subsequently recombine these genes into various Gateway destination vectors carrying the half-YFP moieties. The first of these destination vectors will be a modification of the plasmid pMSP1 designed in our laboratory (SBG). pMSP1 is a transient expression vector based upon pUC118 and contains the "super-promoter", followed by numerous restriction endonuclease sites found in the pBlueScript multiple cloning site (MCS), followed by an ags polyA addition signal. The super-promoter is a strong, constitutive promoter (60) that works especially well in Arabidopsis and tobacco. We and others have used this promoter for high-level expression of genes in plants (58). Protein partners that interact in protoplasts using transient expression assays will also be tested for interaction in stably transformed Arabidopsis plants, using the super-promoter to drive expression of the relevant gene pairs cloned into modified pMSP1-based T-DNA binary vectors generated by placing into their MCS region Gateway attR recombination sites. Additionally, we shall express selected protein fusions from their native plant promoters (see below) cloned into promoterless Gateway T-DNA binary vectors generated by one of us (VC) in his current NSF 2010 award.
2. Gateway destination vectors
The Gateway system (Invitrogen) is based on bacteriophage ? site-specific recombination (49). Gateway cloning introduces the tested gene into the entry vector, pDONR207 (Invitrogen), by in vitro recombination between the attB1 and attB2 sequences engineered to flank the tested gene and the attP1 and attP2 sequences, respectively, of pDONR207. This attB x attP recombination is mediated by the BP reaction (Invitrogen) and produces the attL1 and attL2 sequences that flank the tested gene within the pDONR vector. From the entry vector, the tested gene is transferred to the destination vectors by in vitro recombination between these flanking attL1 and attL2 sequences and the attR1 and attR2 sequences, respectively, of the destination vectors (49, see also www.invitrogen.com). This attL x attR recombination is mediated by the LR reaction (Invitrogen) and produces the attB1 and attB2 sequences that flank the tested gene within the destination vector.
Three plasmids constructed in our lab (SBG), pMSP1, pMSP1-kan, and pMSP1-hyg, each carry a "super-promoter" composed of three octopine synthase gene activators and an activator/promoter of the mannopine synthase gene. In addition, pMSP1-kan carries a kanamycin resistance marker for selection of transgenic plants whereas pMSP1-hyg carries a hygromycin resistance marker. We shall convert these vectors into Gateway destination vectors by subcloning the Gateway conversion cassette C.1 (attR1-Cmr-ccdB-attR2, Invitrogen) into their SmaI site. The resulting Gateway destination vectors, designated pMSP1-GW, pMSP1-kan-GW, and pMSP1-hyg-GW, have the following relevant structures:
pMSP1-GW:
"Super-promoter"->
XhoI-
SalI-
HindIII-attR1-Cm
r-
ccdB-attR2-
SpeI-
XbaI-
SacI-
ags polyA/terminator,
in a pUC118 backbone.
pMSP1-kan-GW:
T-DNA right border-"super-promoter" ->
XhoI-
SalI-
HindIII-attR1-Cm
r-
ccdB-attR2-
SpeI-
XbaI-
SacI-
agspolyA/terminator-p
nos-
nptII-BamHI-Ag7polyA/terminator-T-DNA
left border.
pMSP1-hyg-GW: T-DNA right border-"super-promoter"-> XhoI- SalI- HindIII-attR1-Cm r-ccdB-attR2- SpeI- XbaI-SacI- agspolyA/terminator-p nos- hpt- BamHI-Ag7polyA/terminator-T-DNA left border.
pMSP1-GW is a small, multi-copy plasmid that is ideal for studies using transient transformation (via transfection of protoplasts, or microprojectile bombardment or agroinfiltration of plant tissues), whereas the binary vectors pMSP1-kan-GW and pMSP1-hyg-GW are suitable for stable gene expression via Agrobacterium-mediated transformation. Furthermore, due to their different selection markers, both Gateway binary vectors can be introduced into the same plant, either by co-transformation or by crossing transgenic plants containing individual transgenes, resulting in double-transgenic plants that express two proteins of interest with their selectable markers. These three Gateway destination vectors, useful for high-throughput transfer of genes of interest for over-expression in planta, will represent the first half of the first deliverable of the proposed project and will be made available to the community through the Arabidopsis Biological Resource Center (ABRC).
Note that all unrecombined Gateway entry and destination vectors should be propagated in the DB3.1 strain of E. coli (Invitrogen) carrying the gyrA462 gene that confers resistance to the lethal ccdB gene. Following Gateway recombination, ccdB is replaced by the tested gene, allowing s election for the recombinant clones in ccdB-sensitive bacterial strains such as DH5?
3. Fluorescent reporter Citrine-YFP
We shall utilize the citrine variant of Yellow Fluorescent Protein (hereafter simply termed YFP) which has enhanced photostability and is much less sensitive to pH and anions, such as chloride, compared to other YFP variants (34). This reduced sensitivity allows detection of proteins with a wide range of targeting specificities.
For bimolecular complementation, the YFP coding sequence will first be dissected into two portions (40), N-terminal (N-YFP, 154 aa) and C-terminal (YFP-C, 84 aa), to which flanking restriction endonuclease recognition sites will be added. N-YFP and YFP-C will be amplified from the pRSETB-Citrine plasmid (33) using the Pfx DNA polymerase (Invitrogen). Four variations of the tags will be constructed: XhoI-ATG-N-YFP- HindIII, SpeI-N-YFP-STOP- SacI, XhoI-ATG-C-YFP- HindIII, and SpeI-C-YFP-STOP- SacI.
4. Generation of Gateway BMFC destination vectors
Next, the three Gateway destination vectors will be converted to the BMFC vectors. To this end, the XhoI-ATG-N-YFP- HindIII and SpeI-N-YFP-STOP- SacI cassettes will be cloned into the corresponding restriction sites of pMSP1-GW and pMSP1-kan-GW, whereas the XhoI-ATG-C-YFP- HindIII and SpeI-C-YFP-STOP-SacI cassettes will be cloned into the corresponding restriction sites of pMSP1-GW and pMSP1-hyg-GW, resulting in 8 different vectors:
pBMFC-N-YFPa: "super-promoter"-> XhoI-ATG-N-YFP-linker- HindIII-attR1-Cm r- ccdB-attR2- SpeI- XbaI- SacI- ags polyA/terminator.
pBMFC-N-YFPa-kan: same as pBMFC-N-YFPa plus T-DNA border and p nos-nptII- BamHI-Ag7 polyA/terminator for transgenic plant selection.
pBMFC-N-YFPb: "super-promoter"-> XhoI- SalI- HindIII-attR1- Cm r- ccdB-attR2- SpeI-linker-N-YFP-STOP- SacI- ags polyA/terminator.
pBMFC-N-YFPb-kan: same as pBMFC-N-YFPb plus T-DNA borders and pnos-nptII-BamHI-Ag7 polyA/terminator for transgenic plant selection when in pMSP1-kan-GW).
pBMFC-C-YFPa: "super-promoter"-> XhoI-ATG-C-YFP- HindIII-attR1-Cm r- ccdB-attR2- SpeI- XbaI- SacI- ags polyA/terminator.
pBMFC-C-YFPa-hyg: same as pBMFC-C-YFPa plus T-DNA borders and p nos-hpt-BamHI-Ag7 polyA/terminator for transgenic plant selection.
pBMFC-C-YFPb: "super-promoter"->XhoI-SalI-HindIII-attR1-Cmr-ccdB-attR2-SpeI-linker-C-YFP-STOP-SacI-ags polyA/terminator.
pBMFC-C-YFPb-hyg: same as pBMFC-C-YFPb plus T-DNA borders and p nos-nptII- BamHI-Ag7 polyA/terminator for transgenic plant selection.
Vectors pBMFC-N-YFPa and pBMFC-C-YFPa will be used to fuse the N-YFP or C-YFP, respectively, to the N-terminus of the tested gene whereas vectors pBMFC-N-YFPb and pBMFC-C-YFPb will be used to fuse the N-YFP or C-YFP, respectively, to the C-terminus of the tested gene. As mentioned above, due to the presence of different plant selection markers, pBMFC-N-YFPa-kan or pBMFC-N-YFPb-kan can be used in combination with pBMFC-C-YFPa-hyg or pBMFC-C-YFPb-hyg to co-express stably two tested proteins in transgenic plants and analyze their interaction and co-localization. This series of eight Gateway BMFC vectors will represent the second half of the first deliverable of our project and will be made available to the community through the ABRC.
5. Production of entry constructions with the tested genes
Each tested gene will be amplified by PCR using the proofreading Pfx DNA polymerase (Invitrogen), which, as all Pfu enzymes, has the lowest error rate of 1.3x10 -6 mutations per base (13), is optimized for high-yield of long fragments (up to 12 kb), and generates blunt-ended fragments, eliminating the need to "polish" the ends of the PCR products. The PCR primers will be composed of two parts: a gene-specific sequence and the Gateway attB1 or attB2 sequence (highlighted):
Forward attB1 primer:
5'-
ACAAGTTTGTACAAAAAAGCAGGCT
+15-25
gene-specific nucleotides-3'
Reverse attB2 primer: 5'- ACCACTTTGTACAAGAAAGCTGGGT +15-25 gene-specific nucleotides-3'
The resulting amplified genes will therefore be flanked by the attB1 and attB2 sequences at their 5'- and 3'-ends, respectively, allowing their direct recombination into the attP1 and attP2 sites, respectively, of the pDONR207 entry vector. This recombination is performed in vitro using the BP reaction and the BP Clonase mix according to the manufacturer's instructions (Invitrogen) and results in the attL sequences flanking the transferred gene of interest.. In our experience, the efficiency of the recombination of PCR products into pDONR207 is 80-90%. All entry vector clones will be verified by DNA sequencing.
6. Gateway transfer of the tested genes into BMFC destination vectors
The tested genes flanked by the attL1 and attL2 sequences at their 5' and 3' ends, respectively, in the pDONR vector will be recombined into the attR1 and attR2 sites of the BMFC destination vectors. This recombination is performed in vitro using the LR reaction and the LR Clonase mix according to the manufacturer's instructions (Invitrogen). In our experience, the efficiency of the recombination from pDONR into destination vectors is 100%.
The recombination into the BMFC destination vectors is designed to produce translational fusions between the tested protein and the N-YFP or C-YFP tags. Thus, for fusions of N-YFP or C-YFP to the N-terminus of the tested gene in the pBMFC-N-YFPa and pBMFC-C-YFPa vectors, the attB2 site between the C-terminus of the tag and the N-terminus of the tested gene comprises the following open reading frame: 5'-ATG-YFP/C-YFP-AC CCA GCT TTC TTG TAC AAA GTG GT-tested gene-STOP-3'.
For fusions of N-YFP or C-YFP to the C-terminus of the tested gene in the pBMFC-N-YFPb and pBMFC-C-YFPb vectors, the attB1 sequence between the C-terminus of the tested gene and the N-terminus of the tag comprises the following open reading frame: 5'-ATG-tested gene-ACA AGT TTG TAC AAA AAA GCA GGC T-N-YFP/C-YFP-STOP-3'.
These ORFs, therefore, will be taken into account when designing forward attB1 and reverse attB2 primers for PCR amplification of the tested genes for their cloning into pDONR207 (see above). In addition, for cloning into pBMFC-N-YFPa and pBMFC-C-YFPa, the STOP codon will be retained in the tested gene, whereas for cloning into pBMFC-N-YFPb and pBMFC-C-YFPb, the STOP codon will be omitted from the tested gene.
Finally, it is important to note that the attB2 and attB1 sequences separating the N- or the C-terminus of the tested protein from the C- or the N-terminus of the tag, respectively, also function to code for peptide linkers that act as flexible tethers that minimize potential folding interference between the tested protein and the half-YFP tag.
In this proposal, we shall introduce members of several Arabidopsis gene families as well as an entire Arabidopsis cDNA library into the Gateway BMFC vectors.
B. Utilization of the BMFC constructions to investigate interactions of plant proteins with Agrobacterium Virulence (Vir) proteins: Proof of concept
Data from the Citovsky and Gelvin laboratories (much of which was obtained under the auspices of our Plant Genome grant 99-75715) indicated that specific Agrobacterium Vir proteins interact with various Arabidopsis proteins in yeast and/or in vitro. These interactions include: VirB2 with BTI1, BTI2, BTI3, and Rab8 GTPase; VirD2 with importin ? and actin; VirE2 with importin ?, actin, and VIP1; VirE3 with importin ??? and VirF with ASK1 and VIP1.
In addition to these interactions, evidence from our and other laboratories has accumulated for the interaction of VirD2 protein with other Arabidopsis proteins, including a type 2C protein phosphatase (Y. Tao and SBG, unpublished), several cyclophilins (4, 18), and a CAK2Ms protein kinase (4). However, the investigation of these interactions is beyond the scope of this proposal. Presently, the roles of these Arabidopsis proteins in Agrobacterium-mediated transformation are inferred from genetic and biochemical experiments. However, the interaction of these proteins in planta, the critical piece of evidence that these bacterial and host cell factors indeed cooperate during the infection process, has not been demonstrated. Also, it is yet unclear which members of these plant multigene families can and actually do interact with the Vir proteins in planta. We shall therefore use BMFC to test these interactions directly in plant cells.
We shall electroporate pair-wise combinations of the BMFC constructions (in pMSP1-GW) generated as described above into tobacco BY-2 protoplasts. We have chosen to use BY-2 cells, rather than Arabidopsis cells, because in our experience the generation and electroporation of BY-2 protoplasts is more efficient than those of Arabidopsis suspension cells. BY-2 protoplast generation and electroporation are routine in the Gelvin laboratory (e.g., 55). After 24 hours, we shall investigate the extent and intracellular localization of YFP fluorescence using epifluorescence microscopy and (where the YFP fluorescence has been observed) by spinning disk confocal microscopy. The requisite microscopes are available to the Gelvin and Staiger laboratories (Purdue University) and the Citovsky laboratory (SUNY Stony Brook). To improve our throughput, we are requesting the purchase of an additional fluorescence stereoscope for the Purdue groups; this microscope will be most useful for observing protein interactions in whole plant tissues (see below), and it is already available in the Citovsky laboratory. As a negative control, we shall utilize protein pairs that are not known to interact, i.e., VirD2 and VIP1, or VirE2 and ASK1. In some instances, we have determined domains of VirB2, VirD2, and VirE2 that are important for interaction with plant proteins in yeast and in vitro (SBG and VC, unpublished). We shall therefore also use BMFC fusions of Vir proteins with deletions of these domains as additional negative controls. As positive controls, we shall use pairs of proteins known to interact in plants (e.g., actin with actin, actin with fimbrin, etc.).
As noted above, for each protein, four different half-YFP tagged "partners" will be constructed (the N-terminal half of YFP at each terminus of the protein, and the C-terminal half of YFP at each terminus of the protein). As a service to the research community, we feel that it is important to provide proteins with the two different BMFC tags at each terminus. However, screening for interacting partners of Vir proteins with all combinations of these tagged variants would be too laborious (i.e., using all possible combinations of tagged protein partners would quadruple the number of interactions that we would need to examine). As a "first pass" to examine interactions, we shall utilize only a subset of the possible protein combinations. If no interaction is detected, we shall use the next BMFC fusion subset.
Simultaneously, we shall generate homozygous transgenic Arabidopsis lines that express tagged VirE2, VirE3, VirF, and VIP1 proteins constitutively. Constitutive expression of VirE2, VirE3, VirF, and VIP1 is not toxic to plant cells (12, 67, 82; VC, unpublished). However, we (SBG) have observed that constitutive expression of VirD2 is toxic. We shall therefore place the gene encoding the tagged VirD2 protein under inducible promoter control (1). In addition to the transgenic plants expressing Vir proteins, we shall deposit in the ABRC seeds of transgenic plants expressing plant proteins as BMFC constructions; see below. This set of constructions constitutively expressing BMFC-tagged Vir proteins and their interacting Arabidopsis proteins and the corresponding transgenic plants represent the first half of the second deliverable of the project, and will be made available through the ABRC.
The experiments described above would indicate whether a particular combination of plant protein and Vir protein CAN interact in a plant cell if given the opportunity. However, these experiments do not indicate whether the plant protein and the Vir protein actually DO interact in cells in a real plant. It is possible that a particular plant gene is not expressed in particular cell types. For example, because Agrobacterium is a soil microorganism, the most "natural" and frequent site of infection and, thus, expression of Vir-interacting plant proteins, is at the base (crown) of the plant, or in roots. Indeed, the root elongation zone is the region of the root most susceptible to Agrobacterium-mediated transformation (89).
Thus, for those plant proteins that show interaction with Vir proteins in suspension culture cells, we shall conduct further analyses in transgenic Arabidopsis plants. We shall place a BMFC tag at the C-terminus of the open reading frame of a genomic clone of the plant gene. We shall accomplish this using the Fluorescent Tagging of Full-Length Proteins (FTFLP) technique developed in a current NSF 2010 project for which one of us (VC) serves as a PI. In this approach, a full-length gene (including ~3 kbp upstream and 1 kbp downstream of the transcript, assumed to contain the native regulatory elements) is amplified from genomic DNA and tagged with a half-YFP by triple-template PCR. Technically, the two parts (5'- and 3'- regions) of the chosen Arabidopsis gene are amplified and then serve as long primers in a Long Flanking Homology (LFH) PCR (85) using the BMFC fragment and each other as overlapping templates (i.e., triple-template reaction). Therefore, the second round of PCR introduces the BMFC tag into the selected site (usually, 10 amino acid residues upstream of the stop codon) within the Arabidopsis gene without the need for conventional cloning. The resulting tagged gene is inserted into a promoterless Agrobacterium T-DNA binary vector using Gateway in vitro recombination technology (see above); these vectors suitable for native gene expression in plants have already been constructed and successfully tested in our (VC) current NSF 2010 project (detailed FTFLP protocols and additional information about this project can be obtained from its internet site, http://aztec.stanford.edu/gfp/). The constructions will be introduced into wild-type Arabidopsis plants by Agrobacterium-mediated transformation using a flower dip protocol (14) modified for high-throughput application (see 43 and http://aztec.stanford.edu/gfp/). Because the plant gene is now under control of its native promoter, native amounts of the proteins should be produced in the "correct" cells and at the "normal" stage of development. Several expressing homozygous lines (5-8 independent transformants) per each tagged gene will be identified for further use.
We shall cross the tagged plant protein-expressing plants (resistant to one antibiotic) with tagged Vir protein expressing plants (resistant to a second antibiotic) and examine the resulting F1 plants for BMFC (i.e., reconstruction of YFP fluorescence) and intracellular localization of the interacting protein pairs, as described above. This BMFC analysis of the transgenic plants will be performed at four different stages of plant development: (i) seedlings (2-4 leaf stage), (ii) mature vegetative phase (6-8-leaf stage), (iii) flowering and reproductive stage (complete inflorescence), and (iv) senescence phase. These stages were chosen because they represent periods in plant development that are distinct and correspond to major changes in morphogenesis, physiology and/or metabolism. Here, we may discover correlations between the potential developmental and/or organ/tissue specificity of these expression patterns and the known susceptibility of different developmental stages and organs/tissues of Arabidopsis to Agrobacterium-mediated transformation (82, 89). For example, because plant proteins that interact with Vir proteins are likely essential for transformation, their expression levels may be higher in susceptible tissues. We shall be particularly interested in interactions in the root elongation zone, the region of the plant most susceptible to Agrobacterium-mediated transformation (89).
All protein-protein interactions discovered in transient and stable BMFC assays will be catalogued and made available to the research community via our internet site. The set of constructions expressing BMFC-tagged full-length plant genes under their native promoters, the corresponding transgenic plants, and the database of protein-protein interactions identified using these BMFC tools represent the second half of the second deliverable of the project, and will be made available through the ABRC (DNA and seed stocks) and on the project web site (data).
C. Generation of BMFC-tagged Arabidopsis cDNA libraries
The work described in this proposal will generate BMFC constructions for several Arabidopsis proteins. However, there will obviously be many proteins of interest to the research community that will not be covered by our direct efforts. Therefore, as a service to the community, we shall generate cDNA libraries that encode Arabidopsis proteins tagged at their N-termini with either the N-terminal half-YFP, or with the C-terminal half-YFP. To represent better the Arabidopsis transriptome, we shall use whole plants as starting material. In addition, because in our experience (CS) many cytoskeletal genes are expressed only in pollen and are under-represented in "whole plant" cDNA libraries, we shall construct a pollen-specific cDNA library. These Arabidopsis BMFC cDNA libraries will be produced using the CloneMining cDNA Library Construction Kit (Invitrogen). In this approach, the generated cDNA fragments contain attB1 and attB2 Gateway recombination sequences at their 5'- and 3'-ends, respectively.
The amplified clones will be size-fractionated to select 500-2000-bp long fragments using BD CHROMA SPIN columns (Clontech); such small to medium-size cDNA fragments are well-known to produce best results in protein-protein interaction screens, e.g., in the yeast two-hybrid assays (40, R. Sternglanz, personal communication to VC). The resulting cDNA library clones flanked by the attB sequences will be recombined into the attP sites of the pDONR207 vector (see above).
Finally the cDNA library will be transferred from pDONR into our Gateway BMFC destination vectors pBMFC-N-YFPa and pBMFC-C-YFPa. We chose to recombine the cDNA library into the BMFC vectors without the T-DNA borders because high-throughput library screens are best performed by transient gene expression from these smaller vectors rather than by production of transgenic plants. Although the super-promoter can express in pollen (36), the level of expression may not be high. For the pollen cDNA library we shall therefore switch the super-promoter for a the Lat52 pollen-specific promoter (79). The pollen cDNA expression libraries can be used for pollen particle bombardment experiments (25). This part of the proposed research will therefore produce two BMFC versions of each cDNA library, all of which will be available to the research community as the third deliverable of this project. These four libraries will represent the first tool for in planta characterization of protein-protein interactions within the Arabidopsis proteome.
D. Validation of the BMFC-tagged cDNA libraries
We shall provide our BMFC cDNA libraries to the research community as a tool to study protein-protein interactions in the Arabidopsis proteome. Although extensive screening for such interactions is beyond the scope of this proposal, we shall validate the libraries before releasing them to the general public. To this end, we shall perform two types of screening of the libraries. First, we shall screen the pollen cDNA libraries by PCR for the presence of pollen-specific cDNAs such as act12 (51). This screening will assure us that cDNAs of interest to this project are represented in our libraries. Second, we shall test-screen the libraries for interactors of the Agrobacterium VirE2, VirD2, and VirF proteins. Earlier in the proposed research, we shall have generated transgenic Arabidopsis plants expressing these proteins fused to the BMFC tag (see above). Here, we shall prepare protoplasts from these plants and transform them by electroporation with each of the BMFC cDNA libraries. [Note that, in the case of VirD2 transgenic plants, the protoplasts will be treated with inducer to activate VirD2 expression (see above).] The transformed protoplasts will be allowed to express the library for 24 hours, and the protoplasts exhibiting BMFC (i.e., YFP fluorescence) resulting from protein interactions will be isolated using a fluorescence-activated cell sorter. Currently, high-throughput techniques allow protoplasting and sorting of ~10 6 Arabidopsis cells in about 1.5 hours (6). cDNA clones will be amplified by PCR from the isolated protoplasts, and their identity will be determined by DNA sequencing. All isolated interacting clones will be confirmed by another round of BMFC. In these experiments, we expect to isolate cDNA clones coding for actin (interactor of VirD2 and VirE2), importins ? (interactors of VirD2 and possibly VirE2), CAK2Ms PK and/or PP2C (interactors of VirD2), VIP1 (interactor of VirE2), or ASK1 (interactor of VirF) (see Figure 1). These experiments should reveal additional interactors of these Vir proteins, complementing our studies with the selected Arabidopsis gene families.
Distribution of Project Resources and Information, and Intellectual Property Issues
Information regarding the availability of project materials, the generation of BMFC vectors, the resulting constructions, and the cDNA libraries, as well as transgenic plant lines and he results of protein interaction studies will be posted on the project web site. The stocks (plasmids, cDNA libraries, bacterial strains, and transgenic Arabidopsis seeds) will be distributed via the ABRC. Importantly, all these resources and deliverables will be made public as soon as their quality control is completed, and our laboratory personnel will be given no priority access to the characterized genes, no matter how novel or unique their function will prove to be. These dissemination arrangements are long-term and will remain in place after the requested NSF support has ended (see attached letter from ABRC Director, Dr. Scholl).
Our BMFC vectors contain the "super-promoter", which has been patented by Purdue University and is licensed by the Biotechnology Research and Development Corporation (BRDC). The BRDC has agreed to grant a research license to any entity wishing to use these vectors. We shall not seek patent protection for any additional materials generated during the course of this project.
The P.I.s consider the encouragement of young people, especially those from under-represented minorities, to enter a career in science an essential component of this project. To that end, we have established a relationship with Dr. Theodore Muth at Brooklyn College. Dr. Muth already has an active collaboration with Dr. Gelvin's laboratory. Dr. Muth will recruit an undergraduate student (either completing their Sophomore or Junior years); special emphasis will be placed on recruiting traditionally under-represented minority students. This student will spend 8-10 weeks at one of our laboratories during the summer learning about the BMFC system and participating in the research activities of this project. The student will also participate in activities conducted by the summer MARC/AIM programs at the respective universities. These activities include information and tutoring about taking the GRE Examinations, writing a curriculum vitae, etc. After the student returns to his/her home college, he/she will continue their research under Dr. Muth's tutelage. Thus, Dr. Muth's laboratory will develop a stable, long-term relationship with those of the P.I.s. The expectation is that students who continue working on a project throughout the academic year will be more likely to remain interested in pursuing a scientific career. Students who participate in this outreach program the summer after their Sophomore year will be encouraged to continue participation during the summer following their Junior year, thus solidifying their commitment both to the project and to a scientific career.
 This work is made possible
by a grant from the National Science Foundation's Arabidopsis 2010 Research
Program
This work is made possible
by a grant from the National Science Foundation's Arabidopsis 2010 Research
Program