
H. Construction of a full-genome CDNA microarray for Synechocystis
(performed in collaboration with Rob Burnap, Oklahoma State University
BMC Genomics 03
1. Characteristics of the Synechocystis sp. PCC6803 Genome| Total Length: | 3573470 bp (circular) |
| Total Number of ORFs: | 3168 ORFs |
| - on direct strand: | 1660 ORFs |
| - on complementary strand: | 1508 ORFs |
| Percent coding sequence: | 87.01% |
| Percent coding intergenic region: | 12.99% |
| Average length of gene: | 981.4 bp |
| ORFs longer than 2000 bp | 2907 |
| ORFs shorter than 2000 bp | 261 |
| %C+G | 47.72 |
2. Global mRNA Expression Profiling
With the growing availability of complete genomic sequences for scores of prokaryotic organisms and for several eukaryotes, efforts have turned to the development of experimental approaches to measure transcription from a global perspective. DNA microarrays have emerged as a particularly effective tool for genome-wide transcript profiling, especially for studies examining eukaryotic organisms. Information on the temporal patterns of accumulation and disappearance of transcripts for specific groups of genes is suggestive of cellular programs orchestrating these changes [3, 10, 17, 18, 21] . Further developments in the biology now combine the power of knock-out mutations of known regulatory loci with microarray analysis to help elucidate the signal circuitry giving rise to programmatic changes in gene expression [16]. The efforts of tabs like that of Blattner [15, 20] and that field have helped make arrays accessible for many bacterial systems.
3. DNA MicroarraysDNA microarray technology uses microscopic, high-density arrays of DNA target elements immobilized to solid surfaces such as microscope slides. The DNA target elements typically represent specific gene sequences or sub-sequences that hybridize to cognate sequences in samples under investigation (see below). There are currently two major types of DNA microarrays: oligonucleotide microarrays and DNA fragment microarrays. Oligonucleotide microarrays generally utilize in situ oligonucleotide synthesis techniques that directly build individual DNA targets on the surface of the array [2]. Such an approach is epitomized by the proprietary manufacturing methods of the Affymetrix Corporation, a successful, but extremely expensive, implementation. The other type of array, DNA fragment microarray, was initially pioneered for yeast by Brown at Stanford University [4] and for E. coli by Blattner at the University of Wisconsin [17]. This approach is technically accessible and now has been accomplished for Synechocystis sp. PCC6803.
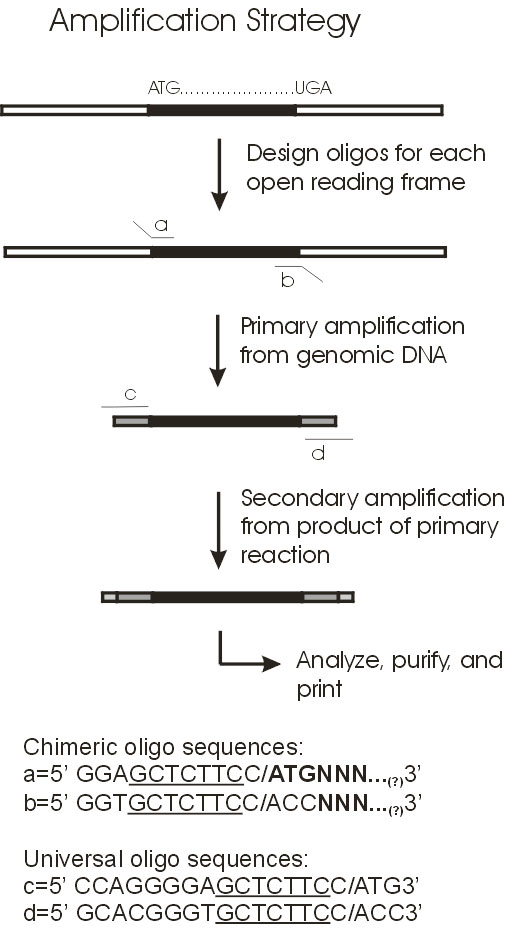
4. Project Design & ExecutionThe primary objective of the project was to construct DNA microarrays for global analysis of transcription in Synechocystis sp. PCC6803. To this end, PCR products for all genes were generated and arrayed onto surface-modified glass slides. For the most part, the PCR gene set was comprised of full-length genes. In instances where the gene is too long to permit efficient PCR amplification in high throughput mode (>2 kb), only the 5' portion was amplified. The amplified genes and gene fragments were flanked by 'adaptamers' that were introduced by utilizing bipartite PCR primers consisting of both gene-specific and engineered sequences [15]. The use of bipartite primers containing both gene-specific and engineered sequences represents a two-stage amplification strategy (Figure 2) that enables several downstream manipulations of the initially amplified gene-set derived from genomic Synechocystis DNA as template. Most importantly, the existence of common adaptamers on every member of the gene set permits subsequent re-amplification of the entire gene set using common (not gene-specific) primers that hybridize to the adaptamers that flank all members of the gene set produced during the initial round of PCR amplification. Thus, the second round of amplification used dilute aliquots of the first round PCR products as template. This affords the ability to generate large amounts of PCR product without excessive depletion of the gene-specific bipartite primer stocks. Additionally, the adaptamers contained restriction sites to facilitate the directional cloning of the PCR products in future studies.
5. PCR primer design considerations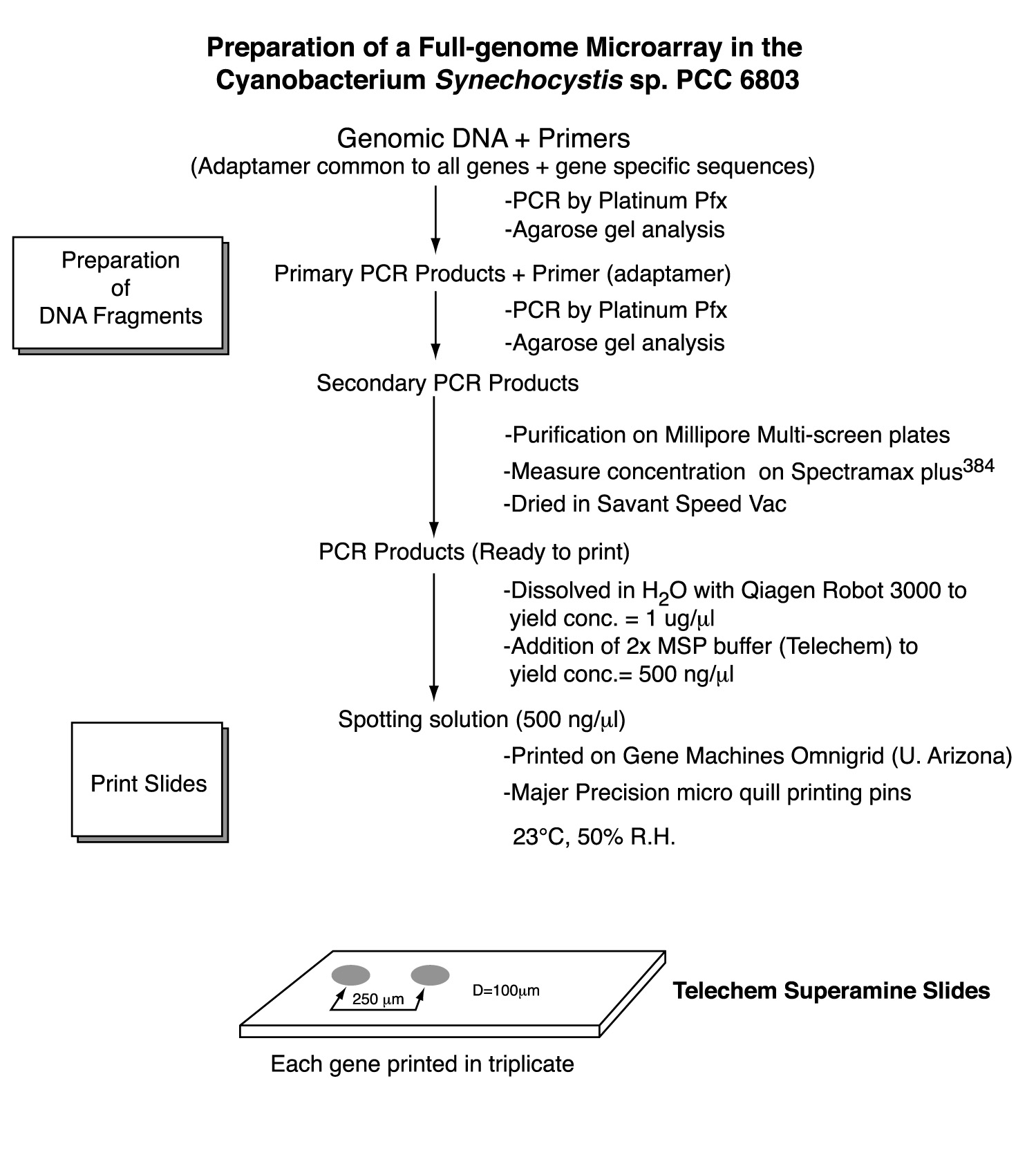{kind=link}
Primers were designed to amplify each of the 3168 genes present in the Synechocystis sp. PCC6803 genome as of May, 2002 [11]. The length of the PCR fragment was limited to 2kb to promote efficient, high-yield amplification of the gene set. Using this criterion, 91% of the ORFs were full length, whereas the remaining 9% were truncated at the 3' ends. The primer design emulate that of the Blattner group [12,17], which incorporates adaptamer sequences appended 5' to gene-specific portion of the bipartite primers. A departure from the Blattner approach will be the modification of the adaptamer sequence to allow re-amplification with a pair of common primers corresponding to the two adaptamer sequences. We used ATG as the start codon for all amplified ORFs irrespective of the actual codon in the genomic sequence, as developed by Blattner. Likewise, TAA terminated every amplified coding sequence, even in the case of the the 3' truncated versions of the long ORFs. These start and stop codons were incorporated into the adaptamer sequences at the 5' and 3' ends of the amplified coding sequence, respectively. The forward adaptamer sequence will consist of 5'-CTTGCTCTTCCATGNNNä.N-3' and the reverse adaptamer sequence 5'-GTTGCTCTTCGTTANNNä.N-3', where N is a stretch of gene-specific nucleotides adding 20-25 gene-specific bases to the 14 base common adaptamer sequence. The length of the gene-specific sequences (NNN..N) were adjusted to achieve a melting temperature in the range of 68-70oC. Sigma-Genosys performed the design and synthesis of the primers and supplied the primers in a 96-well format as pairs of 96 forward and 96 reverse primers for each 96 ORF sequences amplified.
{kind=link}
A portion of the adaptamer sequence corresponds to the non-palindromic 7-base recognition sequence (GCTCTTC) for the restriction enzyme SapI, which is a Type-IIS enzyme with an asymmetric recognition sequence and a cut site starting one base from the 3' end of the recognition sequence and which leaves a 3-base 5' overhang upon cleavage. Introduction of the SapI sites in the adaptamers at the 5' and 3' ends of the coding sequence, together with allowances to preserve the open reading frame, allows for subsequent directional cloning and heterologous expression of the amplified genes in future studies.
6. Amplification of gene setsPCR amplification of the Synechocystis sp. PCC6803 gene-set was performed in two stages. The objective of the proposed two-stage approach was to maximize the yield of PCR products and to avoid depletion of the original stocks of bipartite PCR oligonucleotides. These stocks will not only be used for amplification of the gene-set, but also for the reverse transcription reaction generating the fluorescent hybridization probe as discussed below. We feel it is important to maximize the yield to ensure that the amount of DNA spotted onto the arrays is always saturating. The first round of PCR generated a high fidelity 'master set' of gene fragments using Synechocystis sp. PCC6803 genomic DNA as a template and the bipartite primers. The PCR reaction was catalyzed by Invitrogen Pfu polymerase for maximizing fidelity. For both stages of amplification, the PCR products were analyzed by gel electrophoresis in high throughput fashion (96-lanes per gel).
7. Probe Generation & Microarray HybridizationMicroarray experiments in prokaryotic organisms cannot utilize the poly(A) tails found in eukaryotic mRNA; thus, alternatives to the highly successful methods employed for generating fluorescent cDNA using oligo-dT to prime the reverse transcription labeling reaction have to be devised. We have had moderate success using random hexamers to prime the reaction. However, it will be necessary to generate fluorescent-labeled cDNA in a manner that avoids labeling from highly abundant stable RNA species (this tends to create a high level of non-specific background fluorescence that diminishes the average signal-to-noise ratio of gene-specific hybridization signals from the elements of the microarray). One alternative approach is to use the gene-specific 3' PCR primers manufactured to make the microarrays to also prime the cDNA reaction generating the fluorescent replica of the mRNA population that is being tested. Our preliminary results suggest that this increases the signal-to-noise ratio several-fold. Thus, we wish to make allowances for the possibility that the users will utilize the bipartite gene-specific primers each time an experiment is performed with the microarrays. This approach also has potential downstream dividends, since the introduction of adaptamer sequences allows for directional cloning of the products as well as comparatively inexpensive modification of the adaptamers themselves during secondary amplifications of the gene-set.
To learn more about this work, follow this link.
Back to Sherman Lab Homepage